
GC-MS, LC-MS/MS, Docking and Molecular Dynamics Approaches to Identify Potential SARS-CoV-2 3-Chymotrypsin-Like Protease Inhibitors from Zingiber officinale Roscoe

 ,
, 

Abstract
:1. Introduction
2. Results
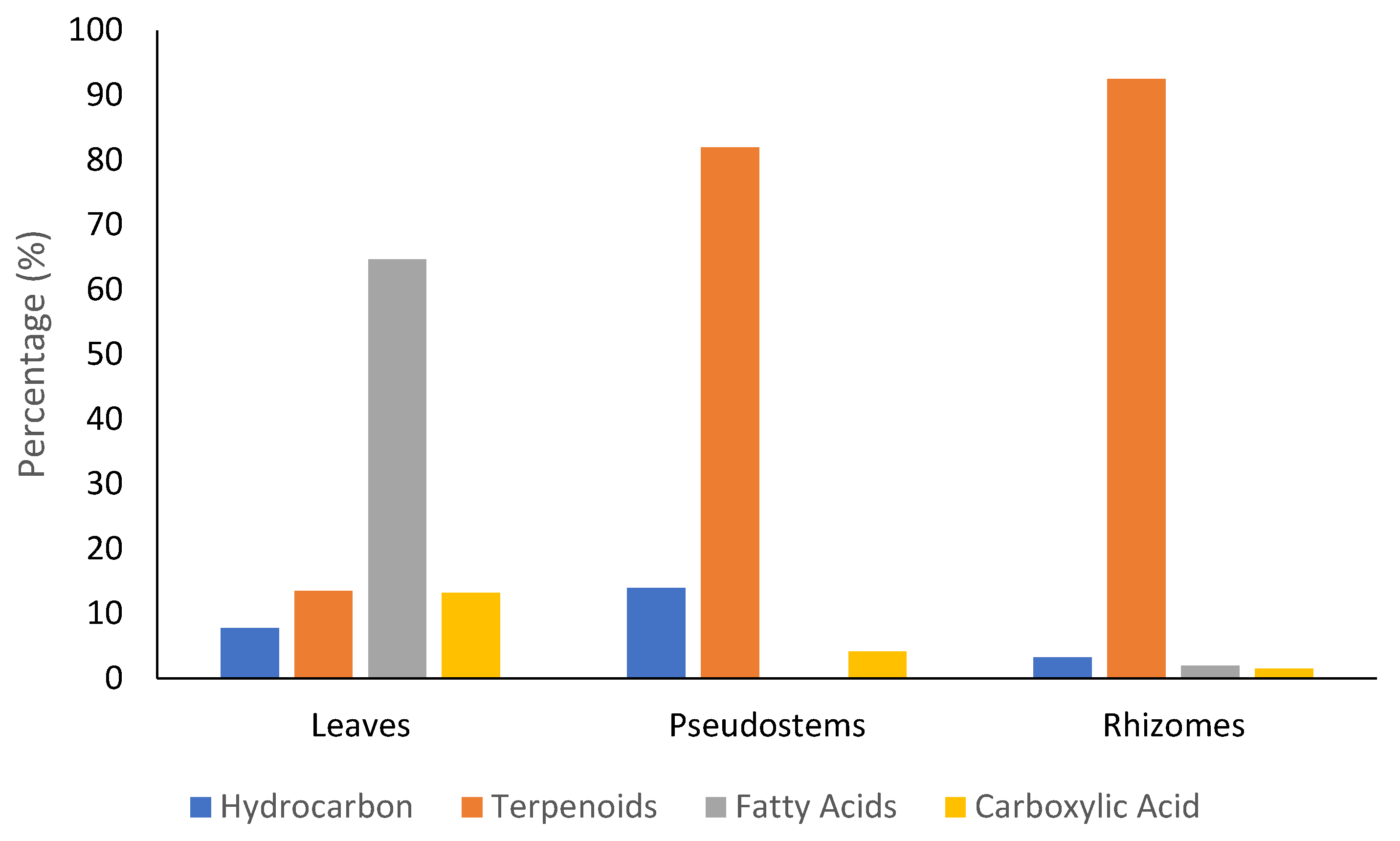
2.1. Extraction and GC-MS Analysis of Z. officinale Methanol Extract
2.2. Isolation and LC-MS/MS Identification of Isolates from Z. officinale n-Hexane Extract
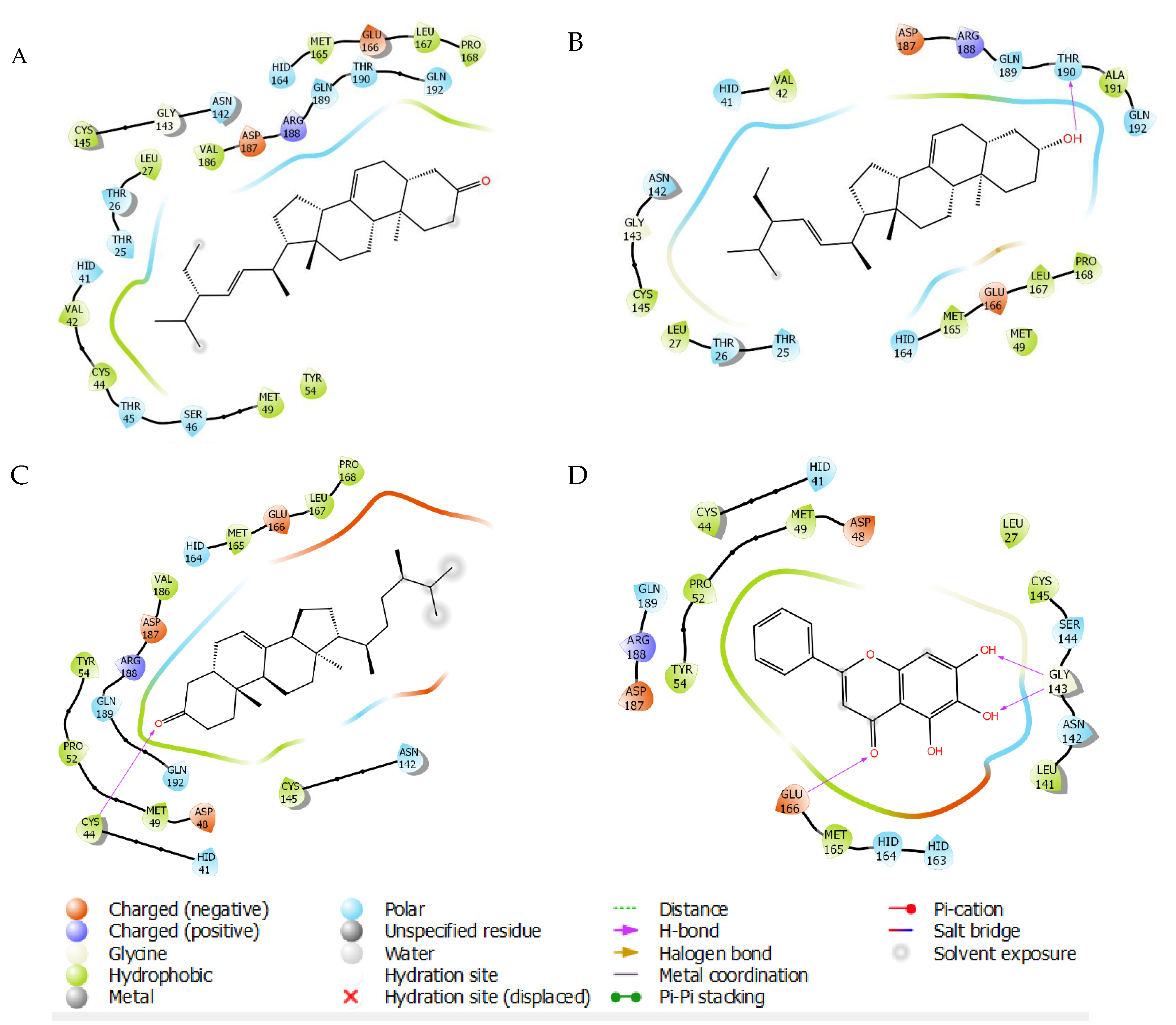
2.3. Molecular Docking
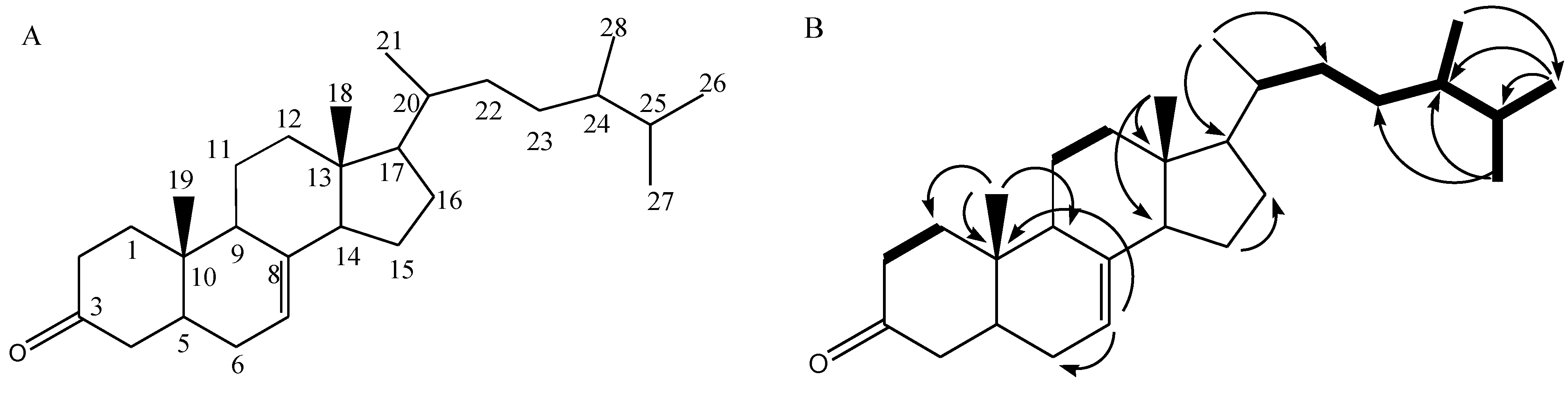
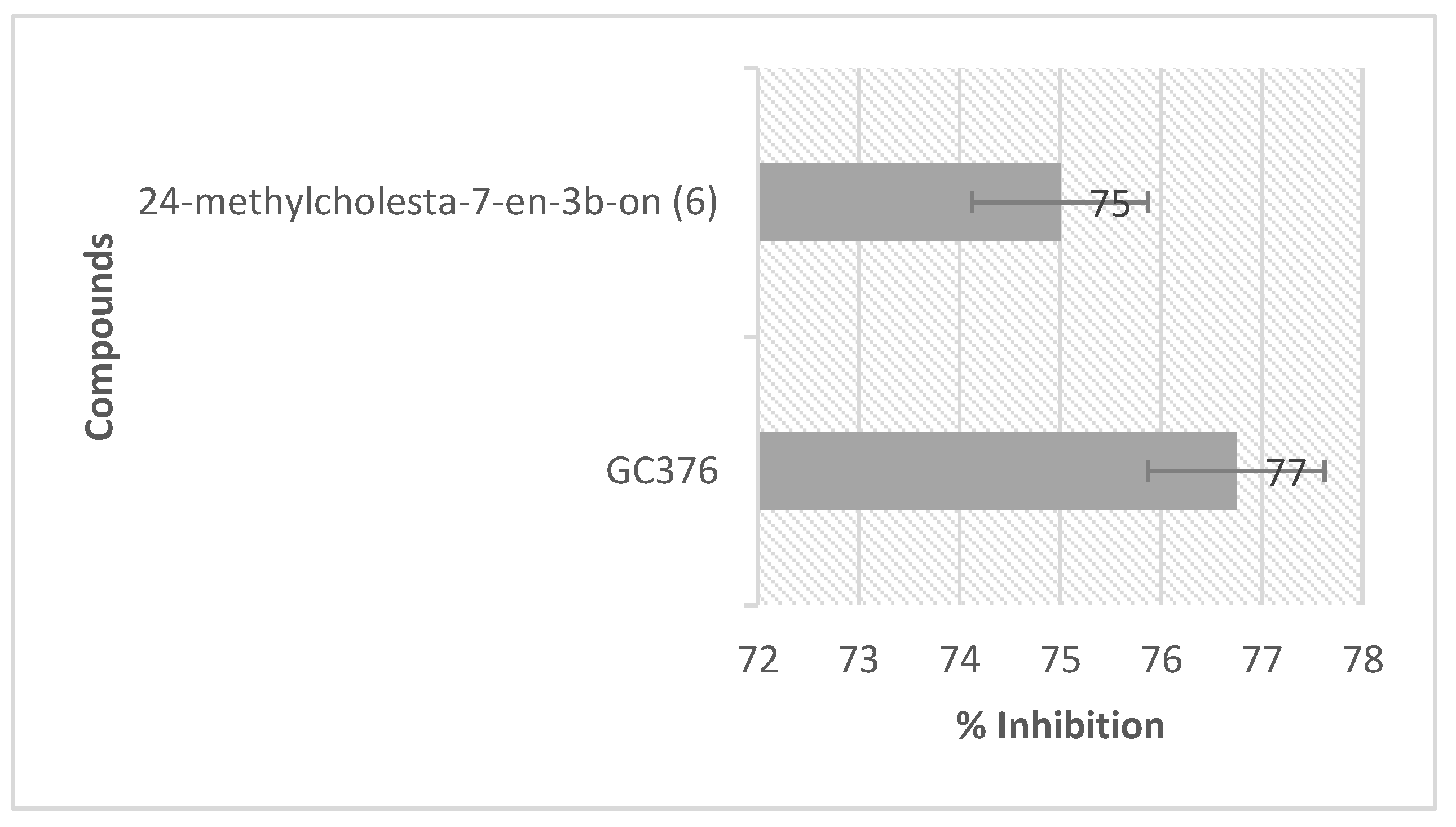
2.4. NMR Analysis and SARS-CoV-2 3CL Protease Inhibitory Activity Verification
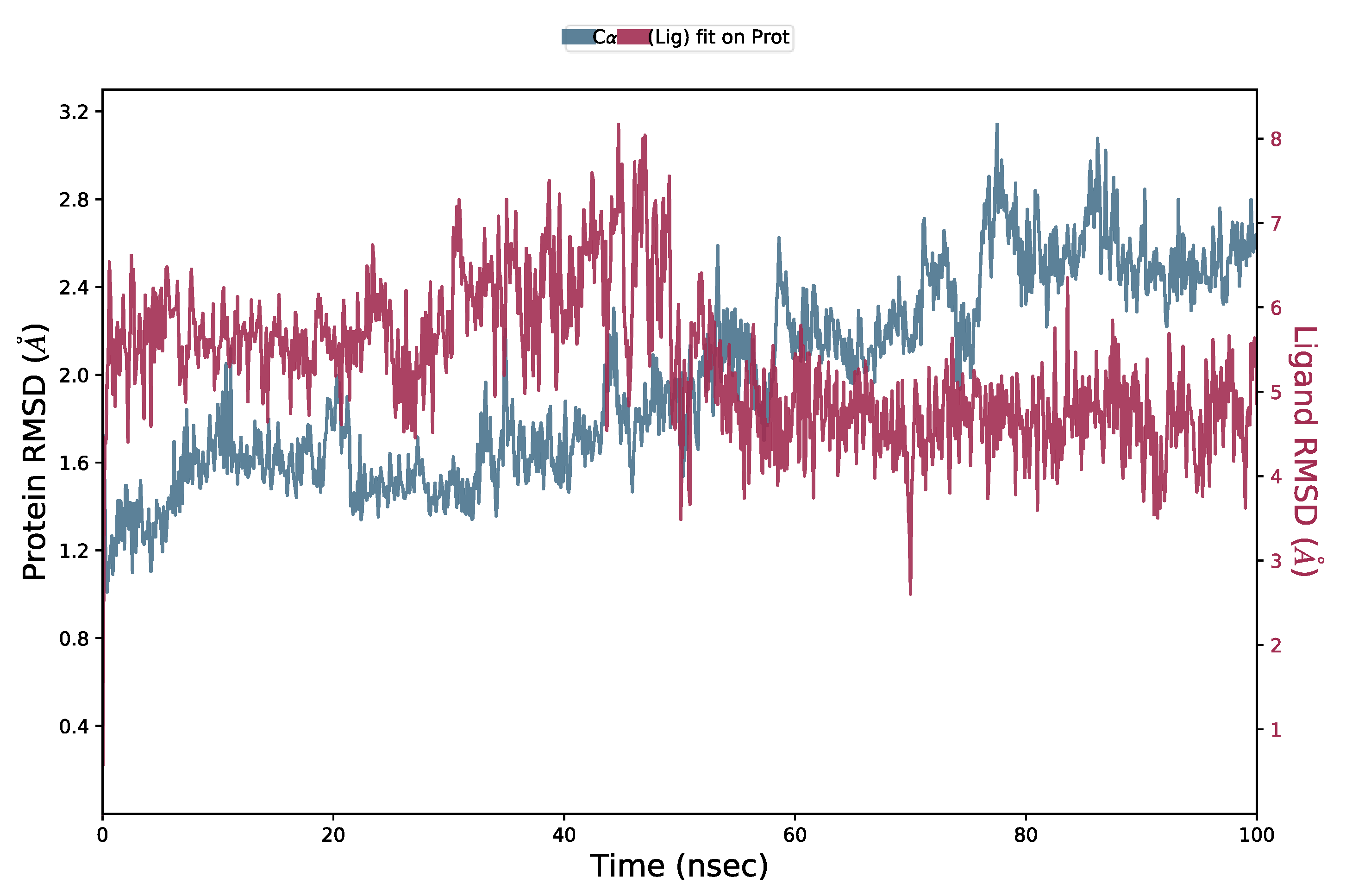
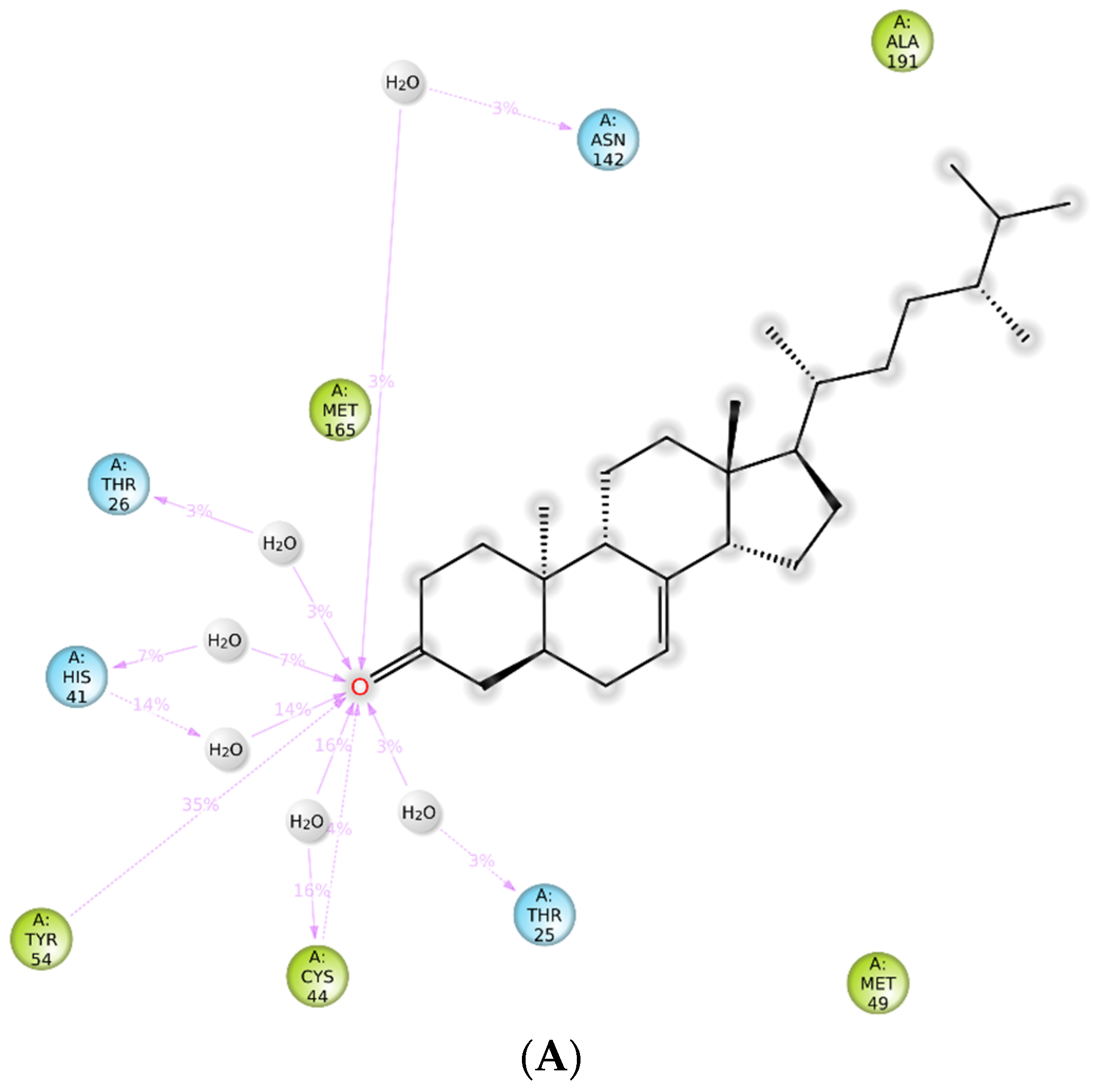
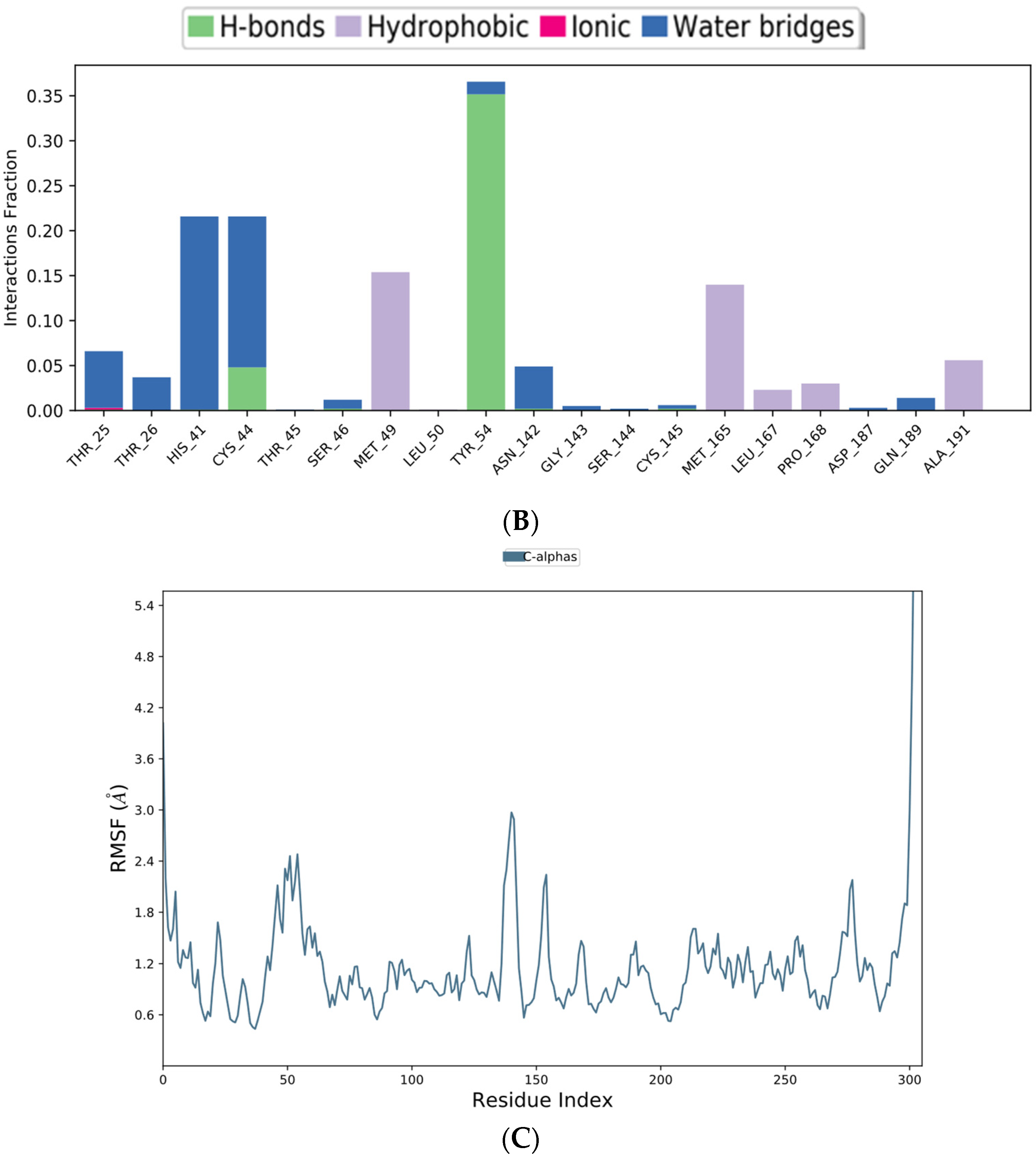
2.5. Molecular Dynamics (MD) Simulation
3. Discussion
4. Materials and Methods
4.1. General
4.2. Plant Material
4.3. Extraction and Isolation
4.4. GC-MS Analysis
4.5. LC-MS/MS Analysis
4.6. Molecular Docking
4.7. SARS-CoV-2 3CL Protease In Vitro Inhibition Assay
4.8. Molecular Dynamics Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, J. SARS-CoV-2: An Emerging Coronavirus that Causes a Global Threat. Int. J. Biol. Sci. 2020, 16, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, S.M.V.; Santos Soares, J.C.; Dos Santos, K.L.; Alves, J.E.F.; Ribeiro, A.G.; Jacob, Í.T.T.; da Silva Ferreira, C.J.; Dos Santos, J.C.; de Oliveira, J.F.; de Carvalho Junior, L.B.; et al. COVID-19 therapy: What weapons do we bring into battle? Bioorg. Med. Chem. 2020, 28, 115757. [Google Scholar] [CrossRef] [PubMed]
- Farooq, S.; Ngaini, Z. Natural and Synthetic Drugs as Potential Treatment for Coronavirus Disease 2019 (COVID-2019). Chem. Afr. 2020, 4, 1–13. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef] [Green Version]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijendra Kumar, N.; Murthy, P.S.; Manjunatha, J.R.; Bettadaiah, B.K. Synthesis and quorum sensing inhibitory activity of key phenolic compounds of ginger and their derivatives. Food Chem. 2014, 159, 451–457. [Google Scholar] [CrossRef]
- Nile, S.; Park, S.W. Chromatographic analysis, antioxidant, anti-inflammatory, and xanthine oxidase inhibitory activities of ginger extracts and its reference compounds. Ind. Crops Prod. 2015, 70, 238–244. [Google Scholar] [CrossRef]
- Citronberg, J.; Bostick, R.; Ahearn, T.; Turgeon, D.K.; Ruffin, M.; Djuric, Z.; Sen, A.; Brenner, D.E.; Zick, S.M. Effects of Ginger Supplementation on Cell-Cycle Biomarkers in the Normal-Appearing Colonic Mucosa of Patients at Increased Risk for Colorectal Cancer: Results from a Pilot, Randomized, and Controlled Trial. Cancer Prev. Res. 2013, 6, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Viennois, E.; Prasad, M.; Zhang, Y.; Wang, L.; Zhang, Z.; Han, M.K.; Xiao, B.; Xu, C.; Srinivasan, S.; et al. Edible ginger-derived nanoparticles: A novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials 2016, 101, 321–340. [Google Scholar] [CrossRef] [Green Version]
- Townsend, E.A.; Siviski, M.E.; Zhang, Y.; Xu, C.; Hoonjan, B.; Emala, C.W. Effects of Ginger and Its Constituents on Airway Smooth Muscle Relaxation and Calcium Regulation. Am. J. Respir. Cell Mol. Biol. 2013, 48, 157–163. [Google Scholar] [CrossRef]
- Wei, C.-K.; Tsai, Y.-H.; Korinek, M.; Hung, P.-H.; El-Shazly, M.; Cheng, Y.-B.; Wu, Y.-C.; Hsieh, T.-J.; Chang, F.-R. 6-Paradol and 6-Shogaol, the Pungent Compounds of Ginger, Promote Glucose Utilization in Adipocytes and Myotubes, and 6-Paradol Reduces Blood Glucose in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2017, 18, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suk, S.; Kwon, G.T.; Lee, E.; Jang, W.J.; Yang, H.; Kim, J.H.; Thimmegowda, N.R.; Chung, M.-Y.; Kwon, J.Y.; Yang, S.; et al. Gingerenone A, a polyphenol present in ginger, suppresses obesity and adipose tissue inflammation in high-fat diet-fed mice. Mol. Nutr. Food Res. 2017, 61, 1700139. [Google Scholar] [CrossRef]
- Akinyemi, A.; Thome, G.R.; Morsch, V.M.; Stefanello, N.; Goularte, J.F.; Belló-Klein, A.; Oboh, G.; Schetinger, M.R.C. Effect of dietary supplementation of ginger and turmeric rhizomes on angiotensin-1 converting enzyme (ACE) and arginase activities in L-NAME induced hypertensive rats. J. Funct. Foods 2015, 17, 792–801. [Google Scholar] [CrossRef]
- Ho, S.-C.; Chang, K.-S.; Lin, C.-C. Anti-neuroinflammatory capacity of fresh ginger is attributed mainly to 10-gingerol. Food Chem. 2013, 141, 3183–3191. [Google Scholar] [CrossRef] [PubMed]
- Rasool, A.; Khan, M.-U.; Ali, M.A.; Anjum, A.A.; Ahmed, I.; Aslam, A.; Mustafa, G.; Masood, S.; Ali, M.A.; Nawaz, M. Anti-avian influenza virus H9N2 activity of aqueous extracts of Zingiber officinalis (Ginger) and Allium sativum (Garlic) in chick embryos. Pak. J. Pharm. Sci. 2017, 30, 1341–1344. [Google Scholar]
- Chang, J.-S.; Wang, K.C.; Yeh, C.F.; Shieh, D.E.; Chiang, L.C. Fresh ginger (Zingiber officinale) has anti-viral activity against human respiratory syncytial virus in human respiratory tract cell lines. J. Ethnopharmacol. 2012, 145, 146–151. [Google Scholar] [CrossRef] [PubMed]
- Ahkam, A.H.; Hermanto, F.E.; Alamsyah, A.; Aliyyah, I.H.; Fatchiyah, F. Virtual prediction of antiviral potential of ginger (Zingiber officinale) bioactive compounds against spike and MPro of SARS-CoV2 protein. Berk. Penelit. Hayati 2020, 25, 52–57. [Google Scholar] [CrossRef]
- Pan, M.; Lei, Q.; Zang, N.; Zhang, H. A Strategy Based on GC-MS/MS, UPLC-MS/MS and Virtual Molecular Docking for Analysis and Prediction of Bioactive Compounds in Eucalyptus Globulus Leaves. Int. J. Mol. Sci. 2019, 20, 3875. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, M.S.; da Silva, V.M.P.; Freitas, L.C.; Silva, S.G.; Cruz, J.N.; Andrade, E.H.D.A. Extraction Yield, Chemical Composition, Preliminary Toxicity of Bignonia nocturna (Bignoniaceae) Essential Oil and in Silico Evaluation of the Interaction. Chem. Biodivers. 2021, 18, e2000982. [Google Scholar] [CrossRef]
- Santana de Oliveira, M.; da Cruz, J.N.; Almeida da Costa, W.; Silva, S.G.; Brito, M.D.P.; de Menezes, S.A.F.; de Jesus Chaves Neto, A.M.; de Aguiar Andrade, E.H.; de Carvalho Junior, R.N. Chemical Composition, Antimicrobial Properties of Siparuna guianensis Essential Oil and a Molecular Docking and Dynamics Molecular Study of its Major Chemical Constituent. Molecules 2020, 25, 3852. [Google Scholar] [CrossRef]
- Hartini, Y.; Saputra, B.; Wahono, B.; Auw, Z.; Indayani, F.; Adelya, L.; Namba, G.; Hariono, M. Biflavonoid as potential 3-chymotrypsin-like protease (3CLpro) inhibitor of SARS-Coronavirus. Results Chem. 2020, 3, 100087. [Google Scholar] [CrossRef] [PubMed]
- Su, H.X.; Yao, S.; Zhao, W.F.; Li, M.J.; Liu, J.; Shang, W.J.; Xie, H.; Ke, C.Q.; Hu, H.C.; Gao, M.N.; et al. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020, 41, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Ong, I.L.H.; Yang, K.-L. Recent developments in protease activity assays and sensors. Analyst 2017, 142, 1867–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Q.-Q.; Xu, X.-Y.; Cao, S.-Y.; Gan, R.-Y.; Corke, H.; Beta, T.; Li, H.-B. Bioactive Compounds and Bioactivities of Ginger (Zingiber officinale Roscoe). Foods 2019, 8, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denaro, M.; Smeriglio, A.; Barreca, D.; De Francesco, C.; Occhiuto, C.; Milano, G.; Trombetta, D. Antiviral activity of plants and their isolated bioactive compounds: An update. Phytother. Res. 2019, 34, 742–768. [Google Scholar] [CrossRef]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Sciortino, M. Antiviral Activity Exerted by Natural Products against Human Viruses. Viruses 2021, 13, 828. [Google Scholar] [CrossRef]
- Xiao, S.; Tian, Z.; Wang, Y.; Si, L.; Zhang, L.; Zhou, D. Recent progress in the antiviral activity and mechanism study of pentacyclic triterpenoids and their derivatives. Med. Res. Rev. 2018, 38, 951–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diniz, L.R.L.; Perez-Castillo, Y.; Elshabrawy, H.A.; Filho, C.; da, S.M.B.; de Sousa, D.P. Bioactive Terpenes and Their Derivatives as Potential SARS-CoV-2 Proteases Inhibitors from Molecular Modeling Studies. Biomolecules 2021, 11, 74. [Google Scholar] [CrossRef]
- Gyebi, G.A.; Ogunro, O.B.; Adegunloye, A.P.; Ogunyemi, O.M.; Afolabi, S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CL pro): An in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2021, 39, 3396–3408. [Google Scholar] [PubMed]
- Loschwitz, J.; Jäckering, A.; Keutmann, M.; Olagunju, M.; Eberle, R.J.; Coronado, M.A.; Olubiyi, O.O.; Strodel, B. Novel inhibitors of the main protease enzyme of SARS-CoV-2 identified via molecular dynamics simulation-guided in vitro assay. Bioorg. Chem. 2021, 111, 104862. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-W.; Tsai, F.-J.; Tsai, C.-H.; Lai, C.-C.; Wan, L.; Ho, T.-Y.; Hsieh, C.-C.; Chao, P.-D.L. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antivir. Res. 2005, 68, 36–42. [Google Scholar] [CrossRef]
- Olubiyi, O.O.; Olagunju, M.; Keutmann, M.; Loschwitz, J.; Strodel, B. High Throughput Virtual Screening to Discover Inhibitors of the Main Protease of the Coronavirus SARS-CoV-2. Molecules 2020, 25, 3193. [Google Scholar] [CrossRef]
- Kac, D.; Barbieri, G.; Falco, M.; Seldes, A.; Gros, E. The major sterols from three species of polyporaceae. Phytochemistry 1984, 23, 2686–2687. [Google Scholar] [CrossRef]
- Y Ragasa, C.; F Galian, R.; Arenal, M.; Tan, V.; Shen, C. Triterpenes and Sterols from Samanea saman. Res. J. Pharm. Biol. Chem. Sci. 2014, 5, 1501–1507. [Google Scholar]
- Meneses-Sagrero, S.E.; Navarro-Navarro, M.; Ruiz-Bustos, E.; Del-Toro-Sánchez, C.L.; Jiménez-Estrada, M.; Robles-Zepeda, R.E. Antiproliferative activity of spinasterol isolated of Stegnosperma halimifolium (Benth, 1844). Saudi Pharm. J. 2017, 25, 1137–1143. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, R.R.; Pise, A.V.; Cheke, R.S.; Shinde, S.D. Recognition of Natural Products as Potential Inhibitors of COVID-19 Main Protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect. 2020, 10, 297–306. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3: Protein Preparation Wizard; Schrödinger, LLC: New York, NY, USA, 2020.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-3: Epik; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-3: Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2020-3: Prime; Schrödinger, LLC: New York, NY, USA, 2020.
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-1: Desmond Molecular Dynamics System; D.E. Shaw Research: New York, NY, USA, 2020.
- Schrödinger Release 2020-1: Maestro Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2020.
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n-Hexane Extract of | Compound’s Code | Compounds Identified | Rt (min) | [M + H]+ |
|---|---|---|---|---|
| Leaves | 1 | (E)-Hexadecyl-ferulate | 9.96 | 419.3156 |
| 2 | Isocyperol | 9.45 | 221.1910 | |
| 3 | N-Isobutyl-(2E,4E)-octadecadienamide | 9.53 | 336.3268 | |
| 4 | Nootkatone | 9.38 | 219.1752 | |
| 5 | Candidate mass C22H45NO | 10.04 | 340.3577 | |
| Pseudostems | 6 | 24-methylcholesta-7-en-3β-on | 10.25 | 399.3618 |
| 7 | Spinasterone | 10.29 | 411.3616 | |
| 8 | Spinasterol | 10.37 | 413.3770 | |
| Rhizomes | 1 | (E)-hexadecyl-ferulate | 9.96 | 419.3156 |
| 2 | Isocyperol | 9.45 | 221.1910 | |
| 3 | N-Isobutyl-(2E,4E)-octadecadienamide | 9.53 | 336.3268 | |
| 4 | Nootkatone | 9.38 | 219.1752 | |
| 9 | 5-hydro-7,8,2′-trimethoxyflavanone | 9.25 | 329.1026 |
| Identified Compounds | Compound’s Code | MMGBSA Binding Energy (kcal/mol) |
|---|---|---|
| Spinasterone | 7 | −87.41 |
| Spinasterol | 8 | −78.11 |
| 24-methylcholesta-7-en-3β-on | 6 | −68.80 |
| N-Isobutyl-(2E,4E)-octadecadienamide | 3 | −65.44 |
| 5-hydro-7,8,2′-trimethoxyflavanone | 9 | −65.42 |
| (E)-hexadecyl-ferulate | 1 | −65.26 |
| Isocyperol | 2 | −62.04 |
| Nootkatone | 4 | −53.24 |
| Baicalein | - | −47.14 |
| Indinavir | - | −76.44 |
| Remdesivir | - | −68.55 |
| P. | δC (m) | δH (m) |
|---|---|---|
| 1 | 35.6 t | 1.65–1.71 (m); 1.98–2.03 (m) |
| 2 | 39.5 t | 1.98–2.03 (m) |
| 3 | 204.6 d | - |
| 4 | 33.9 t | 2.26–2.31 (m, 1H); 2.40–2.42 (m, 1H) |
| 5 | 55.8 t | 1.00–1.02 (m) |
| 6 | 32.9 t | 1.27–1.28 (d); 2.23–2.24 (d) |
| 7 | 123.7 d | 5.71 (s, 1H) |
| 8 | 140.9 s | - |
| 9 | 53.8 d | 0.95 (m) |
| 10 | 38.6 s | - |
| 11 | 20.9 t | 1.00–1.01 (d); 1.52–1.53 (d) |
| 12 | 32.0 t | 1.81–1.84 (m) |
| 13 | 42.2 s | - |
| 14 | 39.6 d | 1.12 (m) |
| 15 | 25.4 t | 1.21 (m) |
| 16 | 29.7 t | 1.27 (m) |
| 17 | 45.8 d | 0.88 (m) |
| 18 | 11.9 q | 0.70 (s,3H) |
| 19 | 17.4 q | 1.17 (s, 3H) |
| 20 | 31.9 d | 1.81–1.84 (m) |
| 21 | 19.8 q | 0.81–0.82 (d, 3H) |
| 22 | 29.1 d | 1.28 (m) |
| 23 | 40.5 t | 1.22 (m, 2H) |
| 24 | 55.9 d | 1.07 (m, 1H) |
| 25 | 36.0 d | 1.24 (m, 1H) |
| 26 | 18.7 q | 0.90–0.91 (d, 3H) |
| 27 | 19.5 q | 1.00–1.01 (d, 3H) |
| 28 | 20.1 q | 0.79–0.82 (d, 3H) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zubair, M.S.; Maulana, S.; Widodo, A.; Pitopang, R.; Arba, M.; Hariono, M. GC-MS, LC-MS/MS, Docking and Molecular Dynamics Approaches to Identify Potential SARS-CoV-2 3-Chymotrypsin-Like Protease Inhibitors from Zingiber officinale Roscoe. Molecules 2021, 26, 5230. https://doi.org/10.3390/molecules26175230
Zubair MS, Maulana S, Widodo A, Pitopang R, Arba M, Hariono M. GC-MS, LC-MS/MS, Docking and Molecular Dynamics Approaches to Identify Potential SARS-CoV-2 3-Chymotrypsin-Like Protease Inhibitors from Zingiber officinale Roscoe. Molecules. 2021; 26(17):5230. https://doi.org/10.3390/molecules26175230
Chicago/Turabian StyleZubair, Muhammad Sulaiman, Saipul Maulana, Agustinus Widodo, Ramadanil Pitopang, Muhammad Arba, and Maywan Hariono. 2021. "GC-MS, LC-MS/MS, Docking and Molecular Dynamics Approaches to Identify Potential SARS-CoV-2 3-Chymotrypsin-Like Protease Inhibitors from Zingiber officinale Roscoe" Molecules 26, no. 17: 5230. https://doi.org/10.3390/molecules26175230
APA StyleZubair, M. S., Maulana, S., Widodo, A., Pitopang, R., Arba, M., & Hariono, M. (2021). GC-MS, LC-MS/MS, Docking and Molecular Dynamics Approaches to Identify Potential SARS-CoV-2 3-Chymotrypsin-Like Protease Inhibitors from Zingiber officinale Roscoe. Molecules, 26(17), 5230. https://doi.org/10.3390/molecules26175230