
Synthesis and Spectroscopic Characterization of Thienopyrazine-Based Fluorophores for Application in Luminescent Solar Concentrators (LSCs)


 ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results
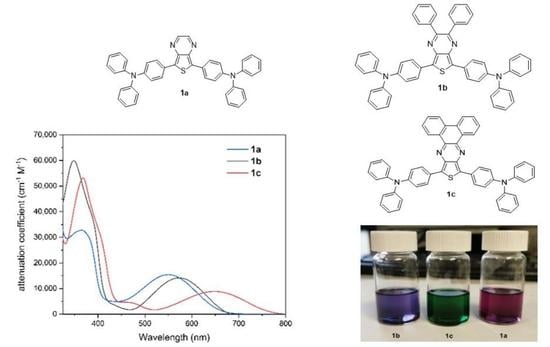
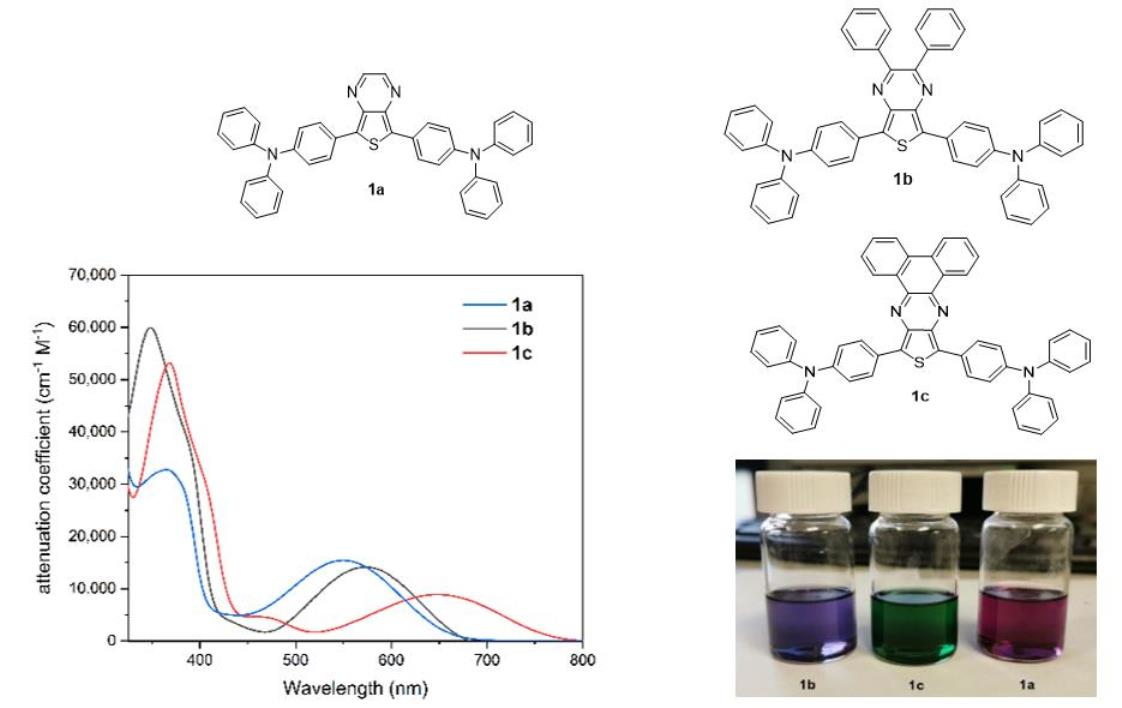
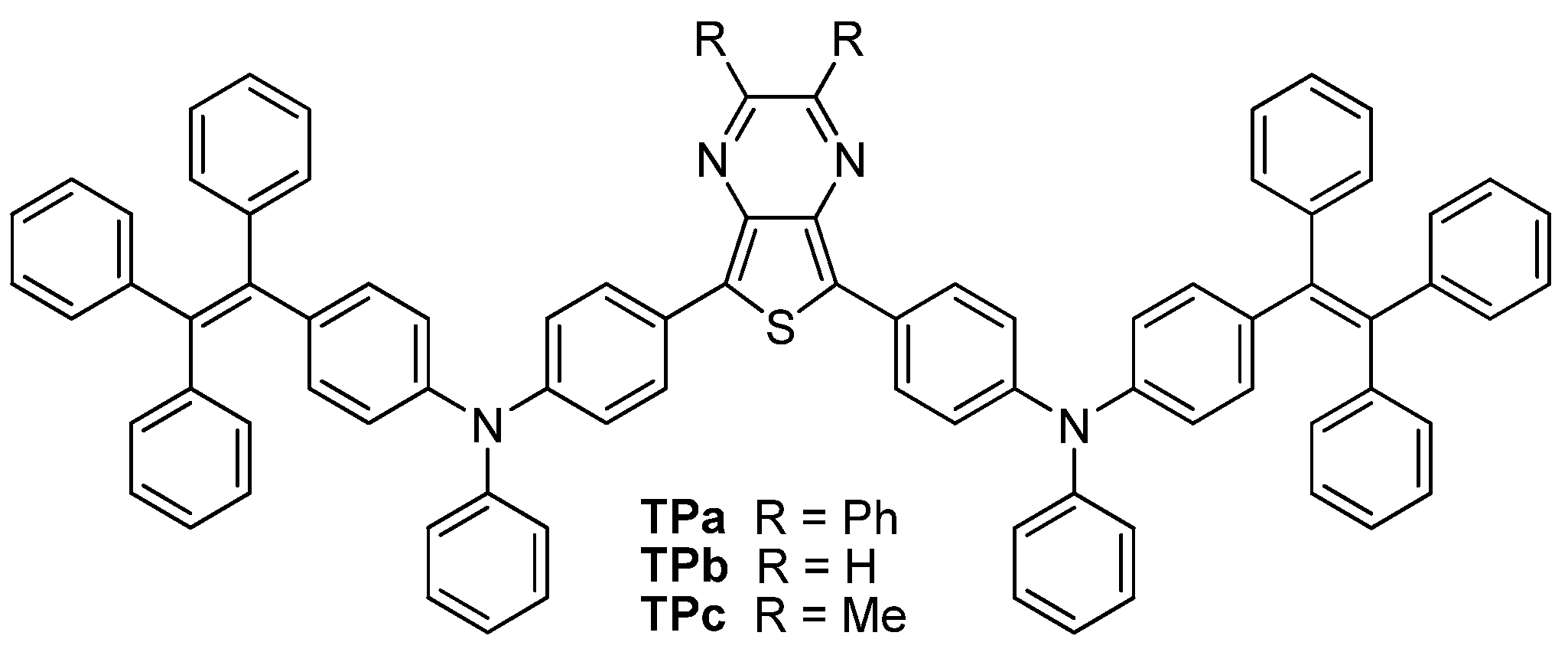
2.1. Synthesis of Dyes
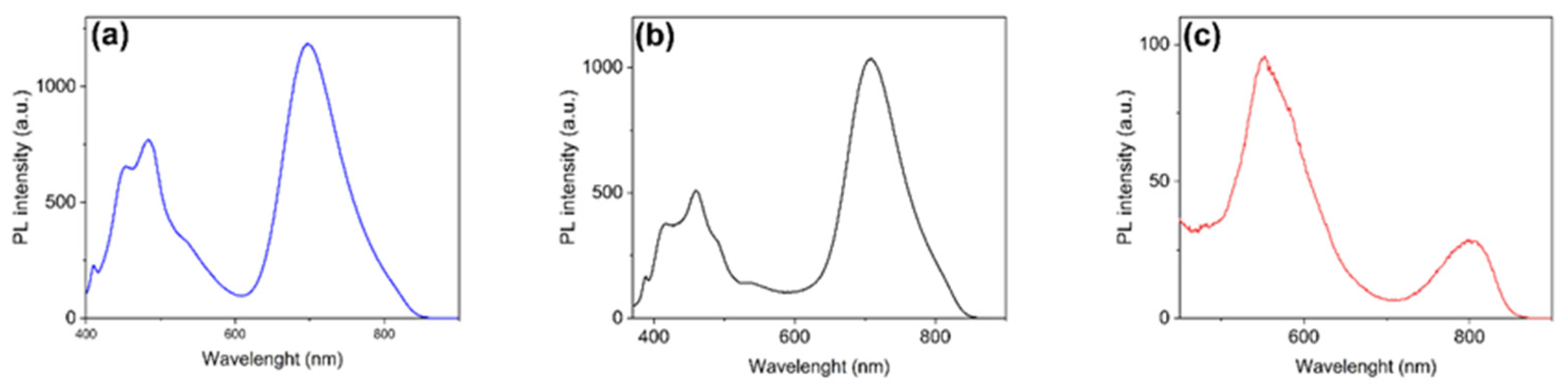
2.2. Photophysical Properties
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Synthesis of 4,4′-(3,4-Dinitrothiophene-2,5-diyl)bis(N,N-diphenylaniline) (6)
3.2.2. General Procedure to Prepare 4,4′-Substituted Thieno[3,4-b]pyrazine-5,7-diyl)bis(N,N-diphenylaniline) (1a–c)
3.2.3. Synthesis of 4,4′-(Thieno[3,4-b]pyrazine-5,7-diyl)bis(N,N-diphenylaniline) (1a)
3.2.4. Synthesis of 4,4′-(2,3-Diphenylthieno[3,4-b]pyrazine-5,7-diyl)bis(N,N-diphenylaniline) (1b)
3.2.5. Synthesis of 4,4′-(Dibenzo[f,h]thieno[3,4-b]quinoxaline-10,12-diyl)bis(N,N-diphenylaniline) (1c)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Armaroli, N.; Balzani, V. Solar electricity and solar fuels: Status and perspectives in the context of the energy transition. Chem.-Eur. J. 2016, 22, 32–57. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, J.-W.; Jung, H.S.; Shin, H.; Park, N.-G. High-efficiency perovskite solar cells. Chem. Rev. 2020, 120, 7867–7918. [Google Scholar] [CrossRef]
- Kokkonen, M.; Talebi, P.; Zhou, J.; Asgari, S.; Soomro, S.A.; Elsehrawy, F.; Halme, J.; Ahmad, S.; Hagfeldt, A.; Hashmi, S.G. Advanced research trends in dye-sensitized solar cells. J. Mater. Chem. A 2021, 9, 10527–10545. [Google Scholar] [CrossRef]
- Inganäs, O. Organic photovoltaics over three decades. Adv. Mater. 2018, 30, 1800388. [Google Scholar] [CrossRef]
- Papakonstantinou, I.; Portnoi, M.; Debije, M.G. The hidden potential of luminescent solar concentrators. Adv. Energy Mater. 2021, 11, 2002883–2002896. [Google Scholar] [CrossRef]
- Meinardi, F.; Bruni, F.; Brovelli, S. Luminescent solar concentrators for building-integrated photovoltaics. Nat. Rev. Mater. 2017, 2, 17072. [Google Scholar] [CrossRef]
- Batchelder, J.S.; Zewai, A.H.; Cole, T. Luminescent solar concentrators 1: Theory of operation and techniques for performance evaluation. Appl. Opt. 1979, 18, 3090. [Google Scholar] [CrossRef]
- Weber, W.H.; Lambe, J. Luminescent greenhouse collector for solar radiation. Appl. Opt. 1976, 15, 2299. [Google Scholar] [CrossRef] [PubMed]
- Roncali, J. Luminescent solar collectors: Quo vadis? Adv. Energy Mater. 2020, 10, 2001907. [Google Scholar] [CrossRef]
- Lu, X.; Fan, S.; Wu, J.; Jia, X.; Wang, Z.; Zhou, G. Controlling the charge transfer in D − A − D chromophores based on pyrazine derivatives. J. Org. Chem. 2014, 79, 6480–6489. [Google Scholar] [CrossRef] [PubMed]
- Goti, G.; Calamante, M.; Coppola, C.; Dessì, A. Donor-acceptor-donor thienopyrazine-based dyes as NIR- emitting AIEgens. Eur. J. Org. Chem. 2021, 2655–2664. [Google Scholar] [CrossRef]
- Zhou, E.; Cong, J.; Yamakawa, S.; Wei, Q.; Nakamura, M.; Tajima, K.; Yang, C.; Hashimoto, K. Synthesis of thieno[3,4-b]pyrazine-based and 2,1,3-benzothiadiazole-based donor-acceptor copolymers and their application in photovoltaic devices. Macromolecules 2010, 43, 2873–2879. [Google Scholar] [CrossRef]
- Ju, X.; Kong, L.; Zhao, J.; Bai, G. Synthesis and electrochemical capacitive performance of thieno [ 3, 4-b ] pyrazine-based Donor-Acceptor type copolymers used as supercapacitor electrode material. Electrochim. Acta 2017, 238, 36–48. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Direct synthesis of CdSe nanocrystals within a conducting metallopolymer: Toward improving charge transfer in hybrid nanomaterials. Chem. Commun. 2016, 52, 13112–13115. [Google Scholar] [CrossRef]
- Wang, W.; Guo, H.; Jones, R.A. N-heterocyclic carbene complexes of Pd(II) and terthiophene backbone. Dalton Trans. 2019, 48, 14440–14449. [Google Scholar] [CrossRef]
- Wu, W.; Yang, J.; Hua, J.; Tang, J.; Zhang, L.; Long, Y.; Tian, H. Efficient and stable dye-sensitized solar cells based on phenothiazine sensitizers with thiophene units. J. Mater. Chem. 2010, 1772–1779. [Google Scholar] [CrossRef]
- Caspar, J.V.; Meyer, T.J. Application of the energy gap law to nonradiative, excited-state decay. J. Phys. Chem. 1983, 87, 952–957. [Google Scholar] [CrossRef]
- McNamara, L.E.; Liyanage, N.; Peddapuram, A.; Murphy, J.S.; Delcamp, J.H.; Hammer, N.I. Donor–Acceptor–Donor thienopyrazines via Pd-catalyzed C–H activation as NIR fluorescent materials. J. Org. Chem. 2016, 81, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ling, Y.; Ding, L.; Xiang, C.; Zhou, G. Quinoxaline-based cross-conjugated luminophores: Charge transfer, piezofluorochromic, and sensing properties. J. Mater. Chem. C 2016, 4, 8496–8505. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Machado, A.E.D.H.; Severino, D.; Ribeiro, J.; De Paula, R.; Gehlen, M.H.; de Oliveira, H.P.M.; Matos, M.D.S.; De Miranda, J.A. Solvent effects on the photophysics of 3-(Benzoxazol-2-yl)-7-(N,N-diethylamino)chromen-2-one. Photochem. Photobiol. Sci. 2004, 3, 79–84. [Google Scholar] [CrossRef]
- Ohta, N. Electric field effects on photochemical dynamics in solid films. Bull. Chem. Soc. Jpn. 2002, 75, 1637–1655. [Google Scholar] [CrossRef]
- Stampor, W.; Mróz, W. Electroabsorption in triphenylamine-based hole-transporting materials for organic light-emitting diodes. Chem. Phys. 2007, 331, 261–269. [Google Scholar] [CrossRef]
- Kasha, M. Characterization of electronic transitions in complex molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Del Valle, J.C.; Catalán, J. Kasha’s rule: A reappraisal. Phys. Chem. Chem. Phys. 2019, 21, 10061–10069. [Google Scholar] [CrossRef]
- Shafikov, M.Z.; Brandl, F.; Dick, B.; Czerwieniec, R. Can coumarins break Kasha’s rule? J. Phys. Chem. Lett. 2019, 10, 6468–6471. [Google Scholar] [CrossRef]
- Kulkarni, A.P.; Zhu, Y.; Jenekhe, S.A. Photodegradation of emissive conjugated copolymers and oligomers containing thienopyrazine. Macromolecules 2008, 41, 339–345. [Google Scholar] [CrossRef]
- Gianfaldoni, F.; De Nisi, F.; Iasilli, G.; Panniello, A.; Fanizza, E.; Striccoli, M.; Ryuse, D.; Shimizu, M. A push-pull silafluorene fluorophore for highly efficient luminescent solar concentrators. RSC Adv. 2017, 7, 37302–37309. [Google Scholar] [CrossRef] [Green Version]
- Carlotti, M.; Fanizza, E.; Panniello, A.; Pucci, A. A fast and effective procedure for the optical efficiency determination of luminescent solar concentrators. Sol. Energy 2015, 119, 452–460. [Google Scholar] [CrossRef]
- Yang, C.; Liu, D.; Lunt, R.R. How to accurately report transparent luminescent solar concentrators. Joule 2019, 3, 2871–2876. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solvent a | λ1abs (nm) | ε1 (cm−1 M−1) | λ2abs (nm) | ε2 (cm−1 M−1) | λ2emi (nm) | Stokes Shift (nm [eV]) | Φf (%) |
|---|---|---|---|---|---|---|---|---|
| 1a | hexane | 365 | 30,100 | 552 | 14,700 | 671 | 119 [0.40] | 6 |
| toluene | 365 | 57,400 | 552 | 15,000 | 698 | 146 [0.47] | 2.6 | |
| THF | 365 | 33,600 | 542 | 15,900 | 709 | 167 [0.54] | <1 | |
| EA | 362 | 34,200 | 540 | 15,700 | 705 | 165 [0.54] | <1 | |
| DCM | 361 | 37,800 | 547 | 15,400 | 731 | 184 [0.57] | <1 | |
| DMSO | 365 | 27,600 | 540 | 12,600 | 737 | 198 [0.61] | <1 | |
| 1b | hexane | 343 | 33,400 | 572 | 8400 | 687 | 115 [0.36] | 3 |
| toluene | 348 | 59,000 | 571 | 15,100 | 708 | 137 [0.42] | <1 | |
| THF | 347 | 58,000 | 570 | 15,000 | 722 | 152 [0.46] | <1 | |
| EA | 345 | 36,700 | 563 | 9100 | 725 | 162 [0.49] | <1 | |
| DCM | 347 | 58,100 | 572 | 15,400 | 736 | 164 [0.48] | <1 | |
| DMSO | 350 | 22,700 | 566 | 5500 | 741 | 175 [0.52] | <1 | |
| 1c | hexane | 364 | 13,000 | 649 | 2000 | 780 | 131 [0.32] | 4 |
| toluene | 368 | 55,200 | 647 | 9500 | 804 | 157 [0.37] | 4 | |
| THF | 367 | 48,000 | 655 | 7600 | 809 | 154 [0.36] | <1 | |
| EA | 365 | 26,100 | 648 | 4000 | 810 | 162 [0.38] | <1 | |
| DCM | 368 | 55,200 | 653 | 8600 | 810 | 157 [0.37] | <1 | |
| DMSO | 372 | 32,900 | 660 | 4700 | 814 | 154 [0.36] | <1 |
| Cmpd. | Conc. (wt%) | λabs (nm) | λems (nm) a | Φf (%) a | Stokes Shift (nm [eV]) a | PLSC (µW) | ηopt (%) b | C |
|---|---|---|---|---|---|---|---|---|
| 1c | 0.4 | 378, 632 | 529 | 16.7 | 151 [0.94] | 65.7 | 6.49 ± 0.5 | 1.07 |
| 0.8 | 377, 632 | 526 | 16.4 | 149 [0.93] | 60.8 | 6.01 ± 0.5 | 1.00 | |
| 1.2 | 372, 632 | 523 | 11.5 | 151 [0.96] | 68.9 | 6.81 ± 0.5 | 1.13 | |
| 1.6 | 372, 636 | 523 | 6.9 | 151 [0.96] | 58.3 | 5.76 ± 0.5 | 0.96 | |
| 2.0 | 376, 632 | 521 | 5.5 | 145 [0.92] | 57.3 | 5.66 ± 0.5 | 0.94 | |
| LR305 | 1.4 | 578 | 613 | 96.0 | 35 [0.13] | 102.2 | 10.1 ± 0.5 | 1.68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yzeiri, X.; Calamante, M.; Dessì, A.; Franchi, D.; Pucci, A.; Ventura, F.; Reginato, G.; Zani, L.; Mordini, A. Synthesis and Spectroscopic Characterization of Thienopyrazine-Based Fluorophores for Application in Luminescent Solar Concentrators (LSCs). Molecules 2021, 26, 5428. https://doi.org/10.3390/molecules26185428
Yzeiri X, Calamante M, Dessì A, Franchi D, Pucci A, Ventura F, Reginato G, Zani L, Mordini A. Synthesis and Spectroscopic Characterization of Thienopyrazine-Based Fluorophores for Application in Luminescent Solar Concentrators (LSCs). Molecules. 2021; 26(18):5428. https://doi.org/10.3390/molecules26185428
Chicago/Turabian StyleYzeiri, Xheila, Massimo Calamante, Alessio Dessì, Daniele Franchi, Andrea Pucci, Francesco Ventura, Gianna Reginato, Lorenzo Zani, and Alessandro Mordini. 2021. "Synthesis and Spectroscopic Characterization of Thienopyrazine-Based Fluorophores for Application in Luminescent Solar Concentrators (LSCs)" Molecules 26, no. 18: 5428. https://doi.org/10.3390/molecules26185428
APA StyleYzeiri, X., Calamante, M., Dessì, A., Franchi, D., Pucci, A., Ventura, F., Reginato, G., Zani, L., & Mordini, A. (2021). Synthesis and Spectroscopic Characterization of Thienopyrazine-Based Fluorophores for Application in Luminescent Solar Concentrators (LSCs). Molecules, 26(18), 5428. https://doi.org/10.3390/molecules26185428