
Interaction of (+)-Strebloside and Its Derivatives with Na+/K+-ATPase and Other Targets

 ,
, 
Abstract
:1. Introduction
2. Results
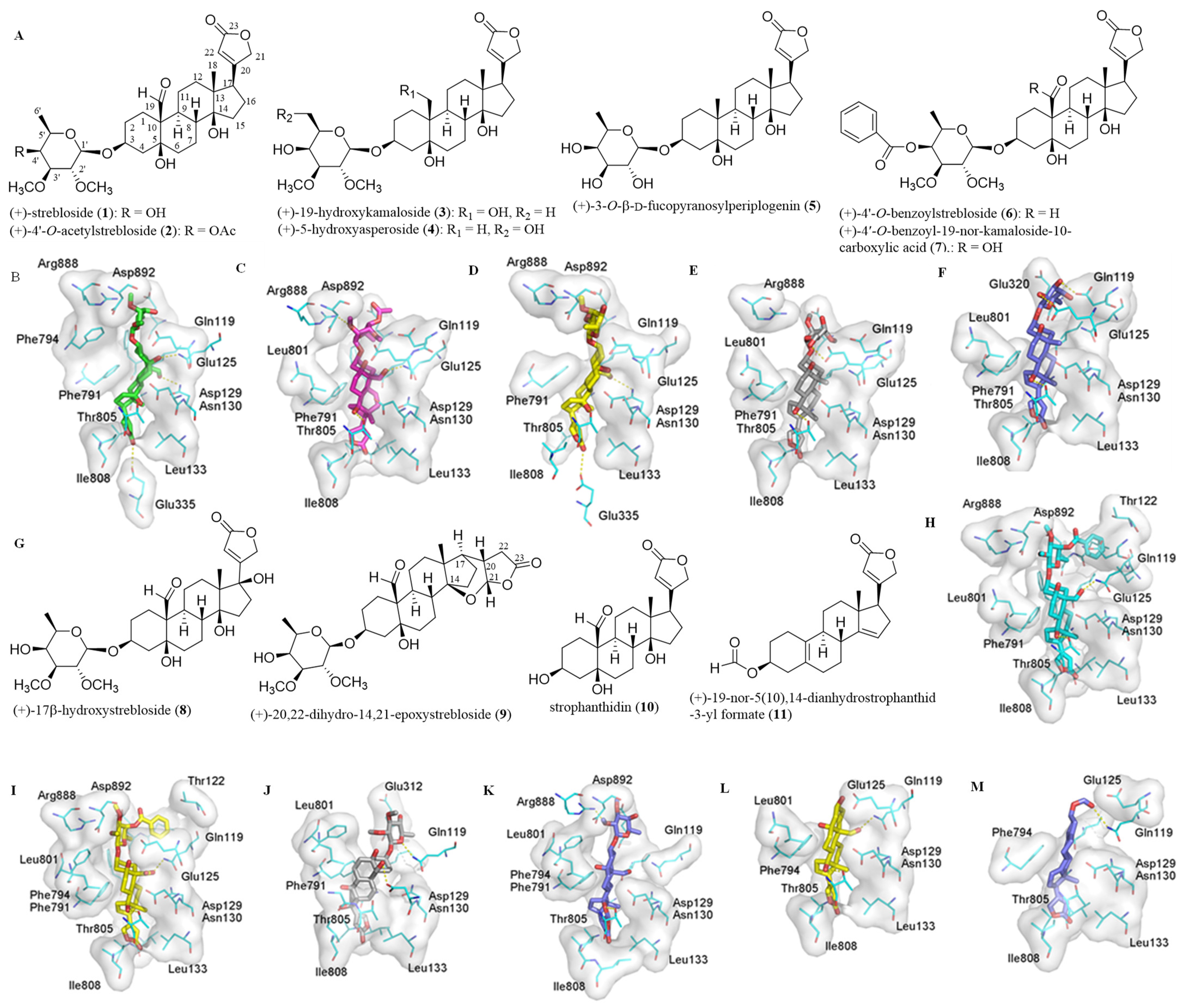
2.1. Impact of the C-3 Saccharide Moiety and the C-10 Formyl Group on the Binding between (+)-Strebloside and NKA
2.2. Impact of the Lactone Unit on the Binding between (+)-Strebloside and NKA
2.3. Binding between the Aglycone of (+)-Strebloside or its Analogue and NKA
2.4. Correlation between Cytotoxicity of 1–11 and the Docking Scores from their Binding to NKA
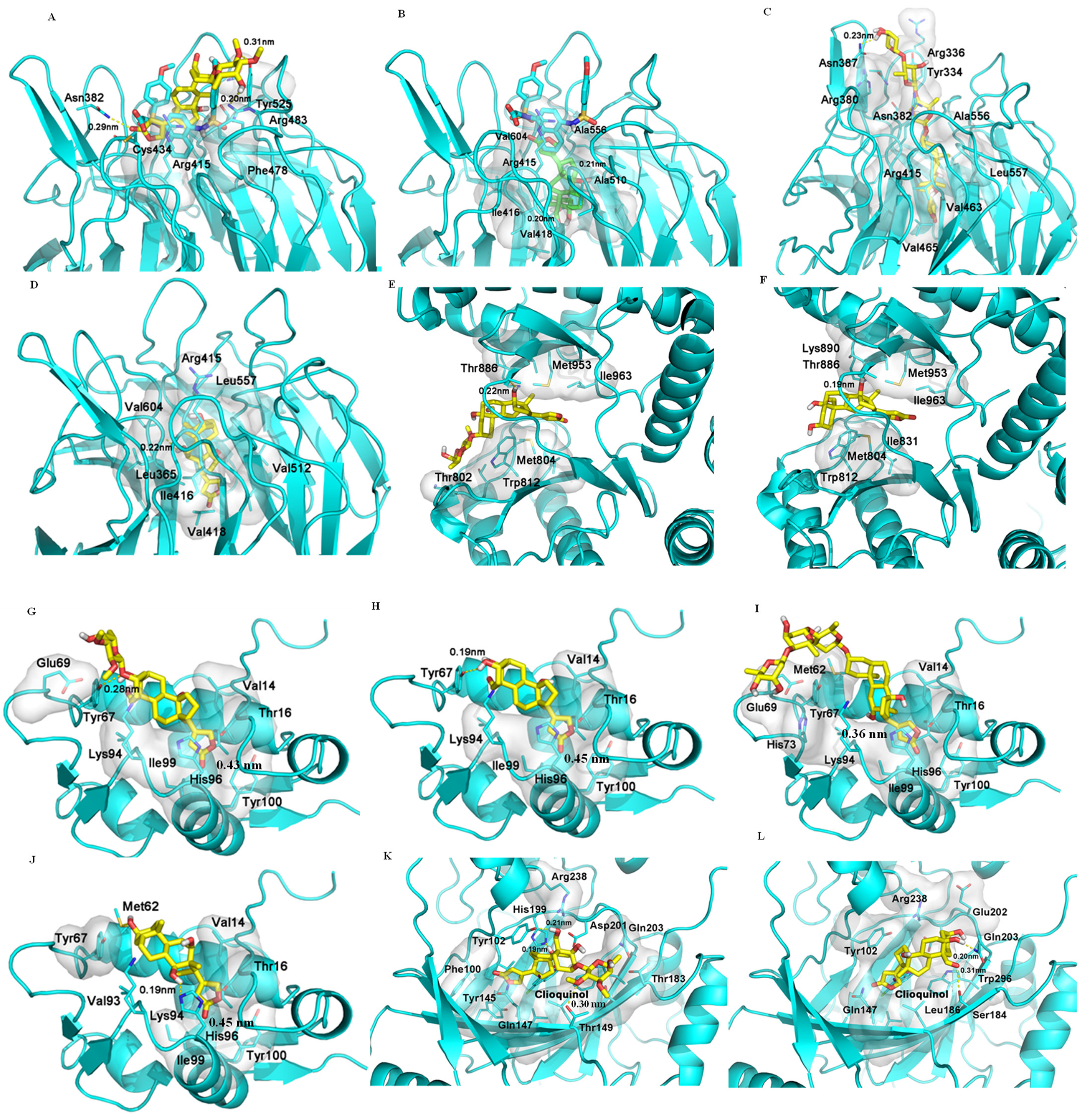
2.5. Binding between (+)-Strebloside (1) or Its Aglycone, Strophanthidin (10), and NF-κB, KEAP1, and PI3K and Akt1
2.6. Binding between (+)-Strebloside (1) or Its Aglycone, Strophanthidin (10) and MDM2
2.7. Binding between (+)-Strebloside (1) or Its Aglycone, Strophanthidin (10), and HDACs
3. Discussion
4. Materials and Methods
4.1. Compounds and Biological Evaluation
4.2. Sequence Alignment and Molecular Modeling for hNKA
4.3. Docking Simulation for NKA
4.4. Molecular Docking for KEAP1, NF-κB, NKA, and PI3K and Akt1
4.5. Molecular Docking for FIH-1, HDAC, and MDM2
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nie, H.; Guan, X.-L.; Li, J.; Zhang, Y.-J.; He, R.J.; Huang, Y.; Liu, B.-M.; Zhou, D.-X.; Deng, S.-P.; Chen, H.-C.; et al. Antimicrobial lignans derived from the roots of Streblus asper. Phytochem. Lett. 2016, 18, 226–231. [Google Scholar] [CrossRef]
- Li, J.; Meng, A.-P.; Guan, X.-L.; Li, J.; Wu, Q.; Deng, S.-P.; Su, X.-J.; Yang, R.-Y. Anti-hepatitis B virus lignans from the roots of Streblus asper. Bioorg. Med. Chem. Lett. 2013, 23, 2238–2244. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.B.S.; Kar, B.; Dolai, N.; Karmakar, I.; Bhattacharya, S.; Haldar, P.K. Antitumor activity and antioxidant status of Streblus asper bark against Dalton’s ascitic lymphoma in mice. Interdiscip. Toxicol. 2015, 8, 125–130. [Google Scholar] [CrossRef] [Green Version]
- Fiebig, M.; Duh, C.-Y.; Pezzuto, J.M.; Kinghorn, A.D.; Farnsworth, N.R. Plant anticancer agents, XLI. Cardiac glycosides from Streblus asper. J. Nat. Prod. 1985, 48, 981–985. [Google Scholar] [CrossRef]
- Miao, D.; Zhang, T.; Xu, J.; Ma, C.; Liu, W.; Kikuchi, T.; Akihisa, T.; Abe, M.; Feng, F.; Zhang, J. Three new cardiac glycosides obtained from the roots of Streblus asper Lour. and their cytotoxic and melanogenesis-inhibitory activities. RSC Adv. 2018, 8, 19570–19579. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.-D.; Zhu, W.-F.; Akihisa, T.; Kikuchi, T.; Ukiya, M.; Maya, F.; Xu, J.; Liu, W.-Y.; Feng, F.; Zhang, J. Cardiac glycosides from the roots of Streblus asper Lour. and their apoptosis-inducing activities in A549 cells. Phytochemistry 2021, 181, 112544. [Google Scholar] [CrossRef]
- Bai, Y.; Zhu, W.; Xu, Y.; Xie, Z.; Akihisa, T.; Manosroi, J.; Sun, H.; Feng, F.; Liu, W.; Zhang, J. Characterization, quantitation, similarity evaluation and combination with Na+/K+-ATPase of cardiac glycosides from Streblus asper. Bioorg. Chem. 2019, 87, 265–275. [Google Scholar] [CrossRef]
- Khare, M.P.; Bhatnagar, S.S.; Schindler, O.; Reichstein, T. Glycosides of Streblus asper. Glycosides and aglycons. Helv. Chim. Acta 1962, 45, 1515–1534. [Google Scholar] [CrossRef]
- Ren, Y.; Chen, W.-L.; Lantvit, D.D.; Sass, E.J.; Shriwas, P.; Ninh, T.N.; Chai, H.-B.; Zhang, X.; Soejarto, D.D.; Chen, X.; et al. Cardiac glycoside constituents of Streblus asper with potential antineoplastic activity. J. Nat. Prod. 2017, 80, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Tan, Q.; Heath, K.; Wu, S.; Wilson, J.R.; Ren, J.; Shriwas, P.; Yuan, C.; Ninh, T.N.; Chai, H.-B.; et al. Cytotoxic and non-cytotoxic cardiac glycosides isolated from the combined flowers, leaves, and twigs of Streblus asper. Bioorg. Med. Chem. 2020, 28, 115301. [Google Scholar] [CrossRef]
- Chen, W.-L.; Ren, Y.; Ren, J.; Erxleben, C.; Johnson, M.E.; Gentile, S.; Kinghorn, A.D.; Swanson, S.M.; Burdette, J.E. (+)-Strebloside-induced cytotoxicity in ovarian cancer cells is mediated through cardiac glycoside signaling networks. J. Nat. Prod. 2017, 80, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Carcache de Blanco, E.J.; Fuchs, J.R.; Soejarto, D.D.; Burdette, J.E.; Swanson, S.M.; Kinghorn, A.D. Potential anticancer agents characterized from selected tropical plants. J. Nat. Prod. 2019, 82, 657–679. [Google Scholar] [CrossRef]
- Diederich, M.; Muller, F.; Cerella, C. Cardiac glycosides: From molecular targets to immunogenic cell death. Biochem. Pharmacol. 2017, 125, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Wu, S.; Burdette, J.E.; Cheng, X.; Kinghorn, A.D. Structural insights into the interactions of digoxin and Na+/K+-ATPase and other targets for the inhibition of cancer cell proliferation. Molecules 2021, 26, 3672. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.-S.; Kuo, S.-C.; Sun, H.-D.; Morris-Natschke, S.L.; Lee, K.-H.; Wu, T.-S. Cytotoxic cardiac glycosides and coumarins from Antiaris toxicaria. Bioorg. Med. Chem. 2014, 22, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Cornelius, F.; Kanai, R.; Toyoshima, C. A structural view on the functional importance of the sugar moiety and steroid hydroxyls of cardiotonic steroids in binding to Na+/K+-ATPase. J. Biol. Chem. 2013, 288, 6602–6616. [Google Scholar] [CrossRef] [Green Version]
- Laursen, M.; Gregersen, J.L.; Yatime, L.; Nissen, P.; Fedosova, N.U. Structures and characterization of digoxin- and bufalin-bound Na+/K+-ATPase compared with the ouabain-bound complex. Proc. Natl. Acad. Sci. USA 2015, 112, 1755–1760. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Shah, V.O.; Ferguson, J.; Hunsaker, L.A.; Deck, L.M.; Vander Jagt, D.L. Cardiac glycosides inhibit LPS-induced activation of pro-inflammatory cytokines in whole blood through an NF-κB-dependent mechanism. Int. J. Appl. Res. Nat. Prod. 2011, 4, 11–19. [Google Scholar]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An overview of Nrf2 signaling pathway and its role in inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- Bhandari, R.; Khanna, G.; Kaushik, D.; Kuhad, A. Divulging the intricacies of crosstalk between NF-κB and Nrf2-KEAP1 pathway in neurological complications of COVID-19. Mol. Neurobiol. 2021, 1–5. [Google Scholar] [CrossRef]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Davies, T.G.; Wixted, W.E.; Coyle, J.E.; Griffiths-Jones, C.; Hearn, K.; McMenamin, R.; Norton, D.; Rich, S.J.; Richardson, C.; Saxty, G.; et al. Monoacidic inhibitors of the Kelch-like ECH-associated protein 1: Nuclear factor erythroid 2-related factor 2 (KEAP1:NRF2) protein-protein interaction with high cell potency identified by fragment-based discovery. J. Med. Chem. 2016, 59, 3991–4006. [Google Scholar] [CrossRef] [PubMed]
- Colarusso, S.; De Simone, D.; Frattarelli, T.; Andreini, M.; Cerretani, M.; Missineo, A.; Moretti, D.; Tambone, S.; Kempf, G.; Augustin, M.; et al. Optimization of linear and cyclic peptide inhibitors of KEAP1-NRF2 protein-protein interaction. Bioorg. Med. Chem. 2020, 28, 115738. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-mediated cellular signaling. Oxidat. Med. Cell. Longev. 2016, 4350965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, H.; Singh, A.; Thapa, K.; Garg, N.; Grewal, A.K.; Singh, T.G. Therapeutic modulation of the phosphatidylinositol 3-kinases (PI3K) pathway in cerebral ischemic injury. Brain Res. 2021, 1761, 147399. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, G.; Zhao, A.; Bondeva, T.; Hirszel, P.; Balla, T. Inhibition of Na+/K+-ATPase activates PI3 kinase and inhibits apoptosis in LLC-PK1 cells. Biochem. Biophys. Res. Commun. 2001, 285, 46–51. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Zhou, Q.; Chen, M.; Zhang, Y.; Liang, H.; Zhao, J.; Zhong, W.; Wang, M. PI3K in cancer: Its structure, activation modes and role in shaping tumor microenvironment. Future Oncol. 2018, 14, 665–674. [Google Scholar] [CrossRef]
- Reddy, D.; Kumavath, R.; Ghosh, P.; Barh, D. Lanatoside C induces G2/M cell cycle arrest and suppresses cancer cell growth by attenuating MAPK, Wnt, JAK-STAT, and PI3K/Akt/mTOR signaling pathways. Biomolecules 2019, 9, 792. [Google Scholar] [CrossRef] [Green Version]
- Reddy, D.; Kumavath, R.; Tan, T.Z.; Ampasala, D.R.; Kumar, A.P. Peruvoside targets apoptosis and autophagy through MAPK Wnt/β-catenin and PI3K/AKT/mTOR signaling pathways in human cancers. Life Sci. 2020, 241, 117147. [Google Scholar] [CrossRef]
- Surget, S.; Khoury, M.P.; Bourdon, J.-C. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. OncoTargets Ther. 2014, 7, 57–68. [Google Scholar]
- Shangary, S.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: A novel approach for cancer therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atatreh, N.; Ghattas, M.A.; Bardaweel, S.K.; Al Rawashdeh, S.; Al Sorkhy, M. Identification of new inhibitors of MDM2-p53 interaction via pharmacophore and structure-based virtual screening. Drug Design Develop. Ther. 2018, 12, 3741–3752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zheng, M.; Li, Z.; Li, R.; Jia, L.; Xiong, X.; Southall, N.; Wang, S.; Xia, M.; Austin, C.P.; et al. Cardiac glycosides inhibit p53 synthesis by a mechanism relieved by Src or MAPK inhibition. Cancer Res. 2009, 69, 6556–6564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Ji, Q.; Dong, L.; Meng, Y.; Xin, G. HDAC4 knockdown induces preeclampsia cell autophagy and apoptosis by miR-29b. Reprod. Sci. 2021, 28, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Sun, S.; Wang, C.; Haas, M.; Yeo, S.; Guan, J.-L. Targeted therapy for mTORC1-driven tumours through HDAC inhibition by exploiting innate vulnerability of mTORC1 hyper-activation. Br. J. Cancer 2020, 122, 1791–1802. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Ribas, H.T.; Heath, K.; Wu, S.; Ren, J.; Shriwas, P.; Chen, X.; Johnson, M.E.; Cheng, X.; Burdette, J.E.; et al. Na+/K+-ATPase-targeted cytotoxicity of (+)-digoxin and several semisynthetic derivatives. J. Nat. Prod. 2020, 83, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.-Y.; Liu, S.-T.; Huang, S.-M.; Chang, Y.-L.; Lin, W.-S. Multiple effects of digoxin on subsets of cancer-associated genes through the alternative splicing pathway. Biochimie 2014, 106, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Zinovkin, R.A.; Grebenchikov, O.A. Transcription factor Nrf2 as a potential therapeutic target for prevention of cytokine storm in COVID-19 patients. Biochemistry 2020, 85, 833–837. [Google Scholar] [PubMed]
- Ramezani, A.; Nahad, M.P.; Faghihloo, E. The role of Nrf2 transcription factor in viral infection. J. Cell. Biochem. 2018, 119, 6366–6382. [Google Scholar] [CrossRef]
- Seelige, R.; Washington, A.; Bui, J.D. The ancient cytokine IL-17D is regulated by Nrf2 and mediates tumor and virus surveillance. Cytokine 2017, 91, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-W.; Chang, H.-Y.; Lee, Y.-Z.; Hsu, H.-Y.; Lee, S.-J. The cardenolide ouabain suppresses coronaviral replication via augmenting a Na+/K+-ATPase-dependent PI3K_PDK1 axis signaling. Toxicol. Appl. Pharmacol. 2018, 356, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Rew, Y.; Sun, D.; Gonzalez-Lopez De Turiso, F.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Deignan, J.; Fox, B.M.; et al. Structure-based design of novel inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2012, 55, 4936–4954. [Google Scholar] [CrossRef]
- Michelsen, K.; Jordan, J.B.; Lewis, J.; Long, A.M.; Yang, E.; Rew, Y.; Zhou, J.; Yakowec, P.; Schnier, P.D.; Huang, X.; et al. Ordering of the N-terminus of human MDM2 by small molecule inhibitors. J. Am. Chem. Soc. 2012, 134, 17059–17067. [Google Scholar] [CrossRef]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef]
- Gupta, R.S.; Chopra, A.; Stetsko, D.K. Cellular basis for the species differences in sensitivity to cardiac glycosides (digitalis). J. Cell. Physiol. 1986, 127, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Montaño, J.M.; Burgos-Morón, E.; López-Lázaro, M. The in vivo antitumor activity of cardiac glycosides in mice xenografted with human cancer cells is probably an experimental artifact. Oncogene 2014, 33, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Azalim, P.; do Monte, F.M.; Rendeiro, M.M.; Liu, X.; O’Doherty, G.A.; Fontes, C.F.; Leitão, S.G.; Quintas, L.E.M.; Noël, F. Conformational states of the pig kidney Na+/K+-ATPase differently affect bufadienolides and cardenolides: A directed structure-activity and structure-kinetics study. Biochem. Pharmacol. 2020, 171, 113679. [Google Scholar] [CrossRef]
- Roth, M.T.; Cardin, D.B.; Borazanci, E.H.; Steinbach, M.; Picozzi, V.J.; Rosemury, A.; Wadlow, R.C.; Newman, R.A.; Berlin, J. A phase II, single-arm, open-label, Bayesian adaptive efficacy and safety study of PBI-05204 in patients with stage IV metastatic pancreatic adenocarcinoma. Oncologist 2020, 25, e1446–e1450. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Kinghorn, A.D. Antitumor potential of the protein phosphatase inhibitor, cantharidin, and selected derivatives. Bioorg. Med. Chem. 2021, 32, 116012. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compd. | Docking Score (kcal/mol) | Cytotoxicity | Compd. | Docking Score (kcal/mol) | Cytotoxicity | ||
|---|---|---|---|---|---|---|---|
| Average | Minimal | Average | Minimal | ||||
| 1 | −10.4 | −11.8 | 0.17 b 0.01 c | 7 | −7.8 | −9.7 | >20 b |
| 2 | −9.7 | −11.5 | 0.47 b | 8 | −8.7 | −11.0 | >20 b |
| 3 | −10.0 | −11.6 | 0.16 b | 9 | −8.8 | −9.8 | >20 b |
| 4 | −9.3 | −10.3 | 0.69 b | 10 | −9.6 | −11.4 | 0.27 c |
| 5 | −10.3 | −11.4 | 0.09 b | 11 | −9.6 | −10.7 | >20 b |
| 6 | −8.8 | −11.0 | 1.2 b | ||||
| Compd. | FIH-1 | KEAP1 | MDM2 | NF-κB p50 | NF-κB p52 | NF-κB p65 | NKA | PI3K |
|---|---|---|---|---|---|---|---|---|
| 1 | −9.3 | −8.9 | −8.3 | −7.4 | −7.0 | −6.9 | −11.8 | −7.0 |
| 10 | −9.9 | −9.7 | −8.1 | −7.2 | −7.1 | −7.1 | −11.4 | −6.9 |
| digoxin | −10.6 | −10.0 | −8.0 | −9.2 | −8.5 | −8.8 | −12.5 | −6.4 |
| digoxigenin | −9.2 | −9.1 | −9.0 | −7.7 | −6.7 | −7.1 | −10.0 | −7.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Y.; Wu, S.; Chen, S.; Burdette, J.E.; Cheng, X.; Kinghorn, A.D. Interaction of (+)-Strebloside and Its Derivatives with Na+/K+-ATPase and Other Targets. Molecules 2021, 26, 5675. https://doi.org/10.3390/molecules26185675
Ren Y, Wu S, Chen S, Burdette JE, Cheng X, Kinghorn AD. Interaction of (+)-Strebloside and Its Derivatives with Na+/K+-ATPase and Other Targets. Molecules. 2021; 26(18):5675. https://doi.org/10.3390/molecules26185675
Chicago/Turabian StyleRen, Yulin, Sijin Wu, Sijie Chen, Joanna E. Burdette, Xiaolin Cheng, and A. Douglas Kinghorn. 2021. "Interaction of (+)-Strebloside and Its Derivatives with Na+/K+-ATPase and Other Targets" Molecules 26, no. 18: 5675. https://doi.org/10.3390/molecules26185675
APA StyleRen, Y., Wu, S., Chen, S., Burdette, J. E., Cheng, X., & Kinghorn, A. D. (2021). Interaction of (+)-Strebloside and Its Derivatives with Na+/K+-ATPase and Other Targets. Molecules, 26(18), 5675. https://doi.org/10.3390/molecules26185675