
Barbeya oleoides Leaves Extracts: In Vitro Carbohydrate Digestive Enzymes Inhibition and Phytochemical Characterization

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
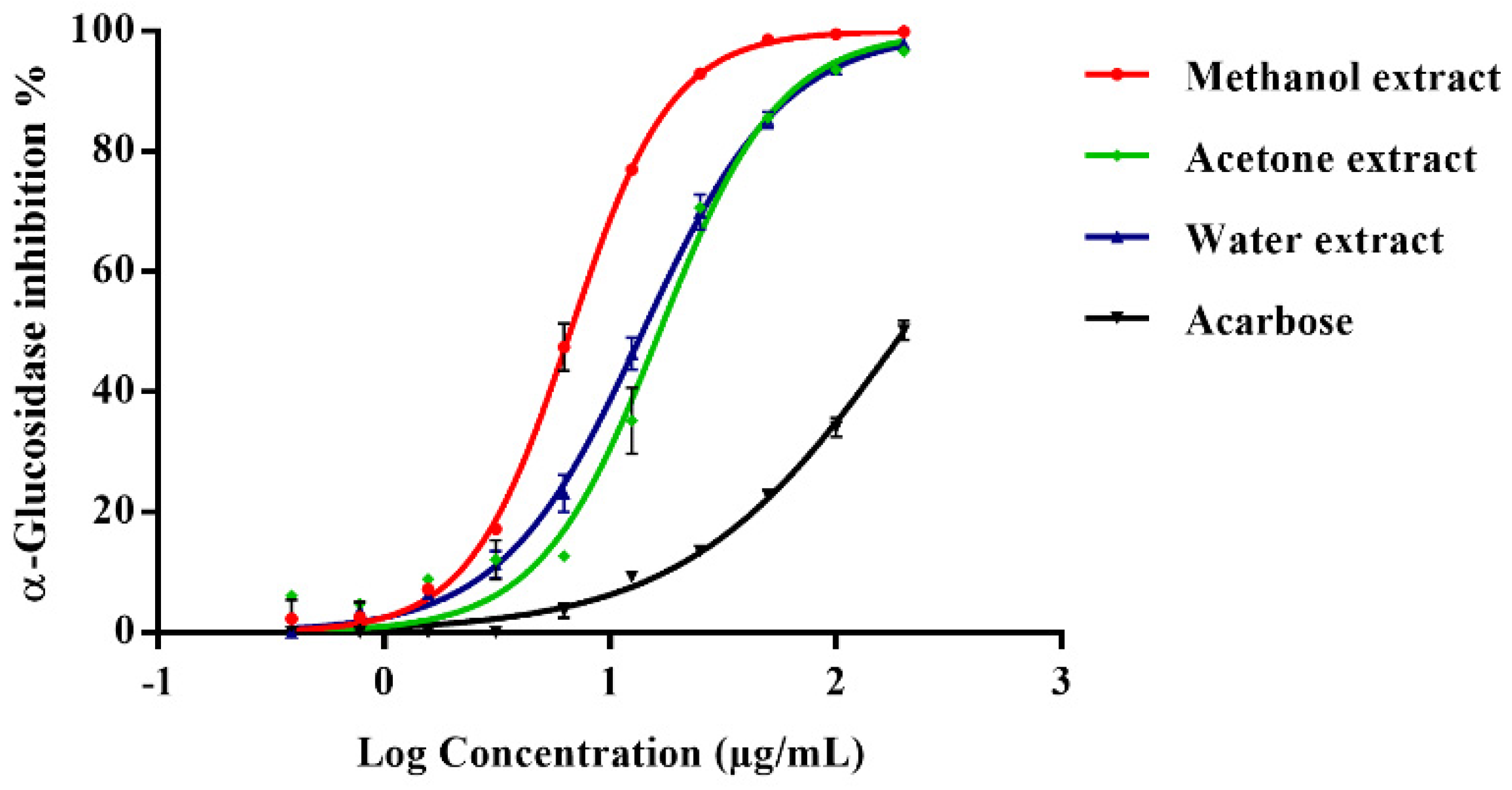
2.1. In Vitro Enzyme Inhibition
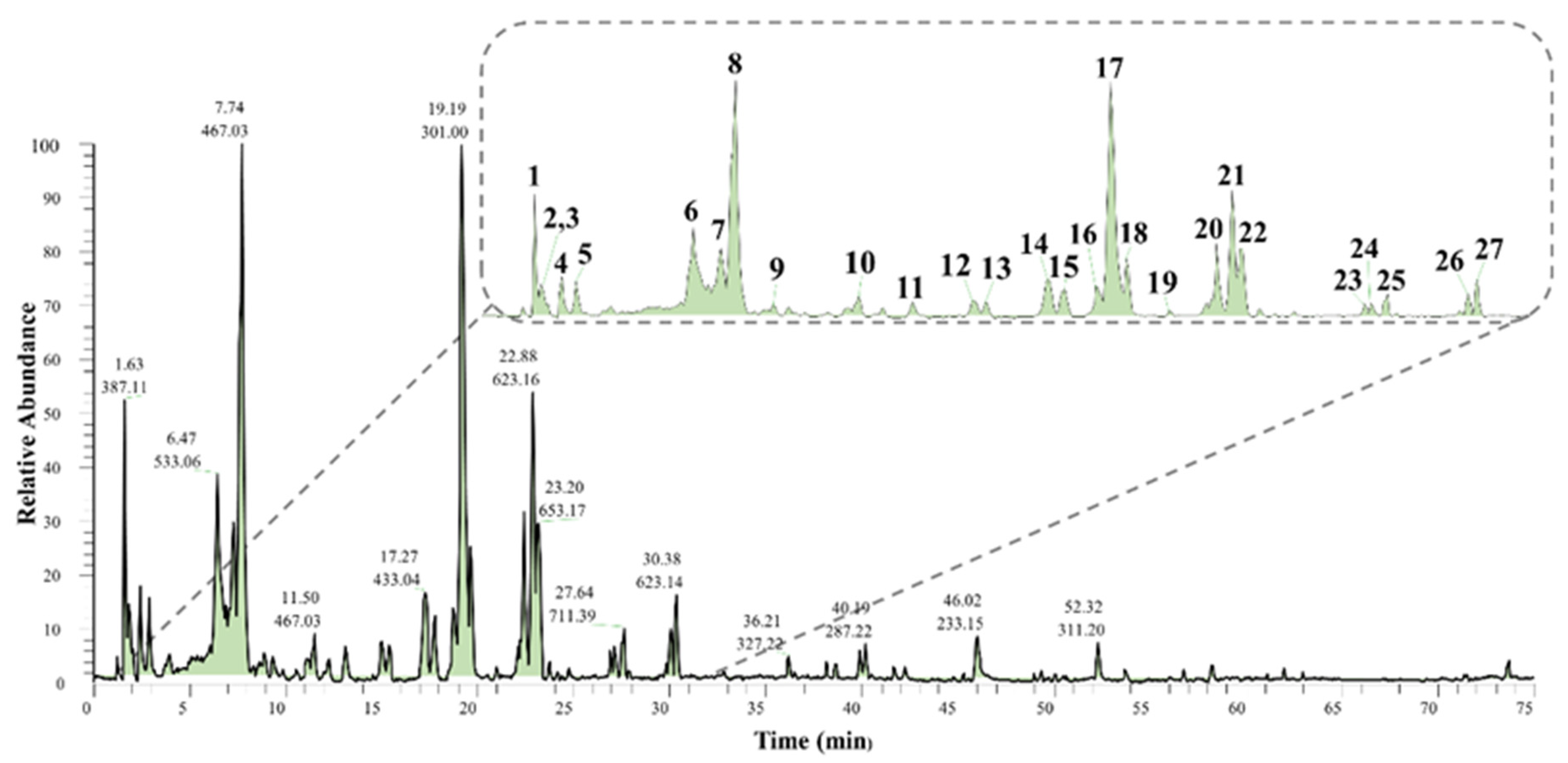
2.2. LC-ESI-MS/MS Profiling
2.2.1. Hydrolysable Tannins and Parent Molecules
2.2.2. Flavonoid Glycosides
2.2.3. Saccharides
2.2.4. Unidentified Compound
2.3. General Discussion
3. Materials and Methods
3.1. Chemical and Reagent
3.2. Plant Material
3.3. Preparation of Plant Extracts
3.4. In Vitro Enzyme Inhibitory Assays
3.4.1. α-Glucosidase Assay
3.4.2. α-Amylase Assay
3.4.3. Half-Maximal Inhibitory Concentration (IC50) Determination
3.4.4. Identification of Aggregate-Based Inhibition
3.4.5. Statistical Analysis
3.5. LC-ESI-MS/MS Analysis of B. oleoides Methanol Extract
3.5.1. Sample Preparation
3.5.2. Instrumentation and Analytical Condition
3.5.3. Data Analysis
3.5.4. Metabolite Identification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Khan, M.A.B.; Hashim, M.J.; King, J.K.; Govender, R.D.; Mustafa, H.; Al Kaabi, J. Epidemiology of type 2 diabetes—Global burden of disease and forecasted trends. J. Epidemiol. Glob. Health 2020, 10, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, A.D. Postprandial hyperglycaemia and α-glucosidase inhibitors. Diabetes Res. Clin. Pract. 1998, 40, 51–55. [Google Scholar] [CrossRef]
- Sachan, A.; Rao, C.; Sachan, N. In vitro studies on the inhibition of α-amylase and α-glucosidase by hydro-ethanolic extract of Pluchea lanceolata, Alhagi pseudalhagi, Caesalpinia bonduc. Pharmacogn. Res. 2019, 11, 310–314. [Google Scholar] [CrossRef]
- Lordan, S.; Smyth, T.J.; Soler-Vila, A.; Stanton, C.; Ross, R.P. The α-amylase and α-glucosidase inhibitory effects of Irish seaweed extracts. Food Chem. 2013, 141, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Rosak, C.; Mertes, G. Critical evaluation of the role of acarbose in the treatment of diabetes: Patient considerations. Diabetes Metab. Syndr. Obes. 2012, 5, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.S.; Kavimani, S.; Jayaveera, K.N. A review on medicinal plants with potential antidiabetic activity. Int. J. Phytopharm. 2011, 2, 53–60. [Google Scholar]
- Yin, Z.; Zhang, W.; Feng, F.; Zhang, Y.; Kang, W. α-Glucosidase inhibitors isolated from medicinal plants. Food Sci. Hum. Wellness 2014, 3, 136–174. [Google Scholar] [CrossRef] [Green Version]
- Sales, P.M.; Souza, P.M.; Simeoni, L.A.; Magalhaes, P.O.; Silveira, D. α-Amylase inhibitors: A review of raw material and isolated compounds from plant source. J. Pharm. Pharm. Sci. 2012, 15, 141–183. [Google Scholar] [CrossRef] [Green Version]
- Arulselvan, P.; Ghofar, H.A.A.; Karthivashan, G.; Halim, M.F.A.; Ghafar, M.S.A.; Fakurazi, S. Antidiabetic therapeutics from natural source: A systematic review. Biomed. Prev. Nutr. 2014, 4, 607–617. [Google Scholar] [CrossRef]
- Ryan, A.J.; Gray, N.M.; Lowe, P.N.; Chung, C.W. Effect of detergent on “promiscuous” inhibitors. J. Med. Chem. 2003, 46, 3448–3451. [Google Scholar] [CrossRef]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; David Phillipson, J.; Greengrass, P.M.; Bowery, N.E.; Cai, Y. Plant polyphenols: Biologically active compounds or non-selective binders to protein? Phytochem. Drug Discov. Today 1997, 44, 441–447. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Screening in a spirit haunted world. Drug Discov. Today 2006, 11, 607–615. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.Y.; Simeonov, A.; Jadhav, A.; Babaoglu, K.; Inglese, J.; Shoichet, B.K.; Austin, C.P. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007, 50, 2385–2390. [Google Scholar] [CrossRef]
- McGovern, S.L.; Shoichet, B.K. Kinase inhibitors: not just for kinases anymore. J. Med. Chem. 2003, 46, 1478–1483. [Google Scholar] [CrossRef]
- Ahmed, B.; Al-Rehaily, A.; Mossa, J. Barbeyol: A new phenolic indane type component from Barbeya oleoides. J. Biosci. 2002, 57, 17–20. [Google Scholar] [CrossRef]
- Al-Oqail, M.; Al-Rehaily, A.; Hassan, W.; Ibrahim, T.; Ahmad, S.; Ebada, S.; Proksch, P. New flavonol glycosides from Barbeya oleoides Schweinfurth. Food Chem. 2012, 132, 2081–2088. [Google Scholar] [CrossRef]
- Li, S.; Lin, Z.; Jiang, H.; Tong, L.; Wang, H.; Chen, S. Rapid identification and assignation of the active ingredients in fufang banbianlian injection using HPLC-DAD-ESI-IT-TOF-MS. J. Chromatogr. Sci. 2016, 54, 1225–1237. [Google Scholar] [CrossRef] [Green Version]
- Cudalbeanu, M.; Ghinea, I.O.; Furdui, B.; Dah-Nouvlessounon, D.; Raclea, R.; Costache, T.; Cucolea, I.E.; Urlan, F.; Dinica, R.M. Exploring new antioxidant and mineral compounds from wild-grown in danube delta biosphere. Molecules 2018, 23, 1247. [Google Scholar] [CrossRef] [Green Version]
- Yisimayili, Z.; Abdulla, R.; Tian, Q.; Wang, Y.; Chen, M.; Sun, Z.; Li, Z.; Liu, F.; Aisa, H.A.; Huang, C. A comprehensive study of pomegranate flowers polyphenols and metabolites in rat biological samples by high-performance liquid chromatography quadrupole time-of-flight mass spectrometry. J. Chromatogr. A 2019, 1604, 460472. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.M.; Koehnlein, E.A.; Bracht, A.; Castoldi, R.; de Morais, G.R.; Baesso, M.L.; Peralta, R.A.; de Souza, C.G.M.; de Sa-Nakanishi, A.B.; Peralta, R.M. Inhibition of salivary and pancreatic α-amylases by a pinhao coat (Araucaria angustifolia) extract rich in condensed tannin. Food Res. Int. 2014, 56, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Bajpai, V.; Kumar, S.; Sharma, K.R.; Kumar, B. Profiling of gallic and ellagic acid derivatives in different plant parts of Terminalia arjuna by HPLC-ESI-QTOF-MS/MS. Nat. Prod. Commun. 2016, 11, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Chernonosov, A.A.; Karpova, E.A.; Lyakh, E.M. Identification of phenolic compounds in Myricaria bracteata leaves by high-performance liquid chromatography with a diode array detector and liquid chromatography with tandem mass spectrometry. Rev. Bras. Farmacogn. 2017, 27, 576–579. [Google Scholar] [CrossRef]
- Zhu, M.; Dong, X.; Guo, M. Phenolic profiling of Duchesnea indica combining Macroporous Resin Chromatography (MRC) with HPLC-ESI-MS/MS and ESI-IT-MS. Molecules 2015, 20, 22463–22475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, N.A.; Rodrigues, E.; Mercadante, A.Z.; de Rosso, V.V. Phenolic compounds and carotenoids from four fruits native from the Brazilian Atlantic Forest. J. Agric. Food. Chem. 2014, 62, 5072–5084. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, L.L.; Vilegas, W.; Dokkedal, A.L. Characterization of flavonoids and phenolic acids in Myrcia bella Cambess. using FIA-ESI-IT-MS(n) and HPLC-PAD-ESI-IT-MS combined with NMR. Molecules 2013, 18, 8402–8416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Johnson, J.V.; Talcott, S.T. Identification of ellagic acid conjugates and other polyphenolics in muscadine grapes by HPLC-ESI-MS. J. Agric. Food. Chem. 2005, 53, 6003–6010. [Google Scholar] [CrossRef] [PubMed]
- Paudel, L.; Wyzgoski, F.J.; Scheerens, J.C.; Chanon, A.M.; Reese, R.N.; Smiljanic, D.; Wesdemiotis, C.; Blakeslee, J.J.; Riedl, K.M.; Rinaldi, P.L. Nonanthocyanin secondary metabolites of black raspberry (Rubus occidentalis L.) fruits: Identification by HPLC-DAD, NMR, HPLC-ESI-MS, and ESI-MS/MS analyses. J. Agric. Food. Chem. 2013, 61, 12032–12043. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Guo, H.; Xu, W.B.; Ge, J.; Li, X.; Alimu, M.; He, D.J. Rapid identification of flavonoid constituents directly from PTP1B inhibitive extract of raspberry (Rubus idaeus L.) leaves by HPLC–ESI–QTOF–MS-MS. J. Chromatogr. Sci. 2016, 54, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yu, H.; Wu, H.; Pan, Y.; Wang, K.; Jin, Y.; Zhang, C. Characterization and quantification by LC-MS/MS of the chemical components of the heating products of the flavonoids extract in pollen typhae for transformation rule exploration. Molecules 2015, 20, 18352–18366. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Shi, P.; Qu, H.; Cheng, Y. Characterization of phenolic compounds in Erigeron breviscapus by liquid chromatography coupled to electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2971–2984. [Google Scholar] [CrossRef]
- Brito, A.; Ramirez, J.E.; Areche, C.; Sepúlveda, B.; Simirgiotis, M.J. HPLC-UV-MS profiles of phenolic compounds and antioxidant activity of fruits from three citrus species consumed in Northern Chile. Molecules 2014, 19, 17400–17421. [Google Scholar] [CrossRef]
- Hofmann, T.; Nebehaj, E.; Albert, L. Antioxidant properties and detailed polyphenol profiling of European hornbeam (Carpinus betulus L.) leaves by multiple antioxidant capacity assays and high-performance liquid chromatography/multistage electrospray mass spectrometry. Ind. Crops Prod. 2016, 87, 340–349. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.S.; Nyberg, N.T.; Staerk, D. Assessment of constituents in Allium by multivariate data analysis, high-resolution α-glucosidase inhibition assay and HPLC-SPE-NMR. Food Chem. 2014, 161, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Olaokun, O.O.; McGaw, L.J.; Eloff, J.N.; Naidoo, V. Evaluation of the inhibition of carbohydrate hydrolysing enzymes, antioxidant activity and polyphenolic content of extracts of ten African Ficus species (Moraceae) used traditionally to treat diabetes. BMC Complement. Altern. Med. 2013, 13, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo-Gonzalez, M.; Reboredo-Rodriguez, P.; Gonzalez-Barreiro, C.; Carrasco-Pancorbo, A.; Cancho-Grande, B.; Simal-Gandara, J. The involvement of phenolic-rich extracts from Galician autochthonous extra-virgin olive oils against the α-glucosidase and α-amylase inhibition. Food Res. Int. 2019, 116, 447–454. [Google Scholar] [CrossRef]
- Oboh, G.; Akinyemi, A.; Ademiluyi, A. Inhibition of α-amylase and α-glucosidase activities by ethanolic extract of Telfairia occidentalis (fluted pumpkin) leaf. Asian Pac. J. Trop. Biomed. 2012, 2, 733–738. [Google Scholar] [CrossRef] [Green Version]
- McDougall, G.J.; Kulkarni, N.N.; Stewart, D. Current developments on the inhibitory effects of berry polyphenols on digestive enzymes. BioFactors 2008, 34, 73–80. [Google Scholar] [CrossRef]
- McDougall, G.J.; Stewart, D. The inhibitory effects of berry polyphenols on digestive enzymes. BioFactors 2005, 23, 189–195. [Google Scholar] [CrossRef]
- Camila Gabriel, K.; de Almeida Gonçalves, G.; Rosely Aparecida, P.; Flavio Augusto Vicente, S.; de Sá-Nakanishi , A.B.; Livia, B.; Jurandir Fernando, C.; Adelar, B.; Rosane Marina, P. Inhibition of α-amylases by condensed and hydrolysable tannins: Focus on kinetics and hypoglycemic actions. Enzym. Res. 2017, 3, 5724902. [Google Scholar] [CrossRef] [Green Version]
- Li, D.Q.; Zhao, J.; Xie, J.; Li, S.P. A novel sample preparation and on-line HPLC–DAD–MS/MS–BCD analysis for rapid screening and characterization of specific enzyme inhibitors in herbal extracts: Case study of α-glucosidase. J. Pharm. Biomed. Anal. 2014, 88, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Trinh, B.T.; Staerk, D.; Jäger, A.K. Screening for potential α-glucosidase and α-amylase inhibitory constituents from selected Vietnamese plants used to treat type 2 diabetes. J. Ethnopharmacol. 2016, 186, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Najari Beidokhti, M.; Andersen, M.V.; Eid, H.M.; Sanchez Villavicencio, M.L.; Staerk, D.; Haddad, P.S.; Jager, A.K. Investigation of antidiabetic potential of Phyllanthus niruri L. using assays for α-glucosidase, muscle glucose transport, liver glucose production, and adipogenesis. Biochem. Biophys. Res. Commun. 2017, 493, 869–874. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Chen, F.; Wang, X.; Jiang, Y.; Lin, S. Anti-diabetic activities of phenolic compounds in muscadine against alpha-glucosidase and pancreatic lipase. Food Sci. Technol. 2012, 46, 164–168. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, X.W.; Chen, X.B.; Wang, Q.X. α-Glucosidase inhibitory constituents from Toona sinensis. Chem. Nat. Compd. 2009, 45, 244–246. [Google Scholar] [CrossRef]
- Wansi, J.D.; Lallemand, M.C.; Chiozem, D.D.; Toze, F.A.A.; Mbaze, L.M.A.; Naharkhan, S.; Iqbal, M.C.; Tillequin, F.; Wandji, J.; Fomum, Z.T. α-Glucosidase inhibitory constituents from stem bark of Terminalia superba (Combretaceae). Phytochemistry 2007, 68, 2096–2100. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Su, A.; Yuan, S.; Zhao, H.; Tan, S.; Hu, C.; Deng, H.; Guo, Y. Evaluation of total flavonoids, myricetin, and quercetin from Hovenia dulcis Thunb. as inhibitors of α-amylase and α-glucosidase. Plant. Foods Hum. Nutr. 2016, 71, 444–449. [Google Scholar] [CrossRef]
- Moradi-Afrapoli, F.; Asghari, B.; Saeidnia, S.; Ajani, Y.; Mirjani, M.; Malmir, M.; Bazaz, R.D.; Hadjiakhoondi, A.; Salehi, P.; Hamburger, M.; et al. In vitro α-glucosidase inhibitory activity of phenolic constituents from aerial parts of Polygonum hyrcanicum. Daru 2012, 20, 37. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.C.; Li, S.L.; Zhang, X.Q.; Ye, W.C.; Zhang, Q.W. Flavonoids with α-glucosidase inhibitory activities and their contents in the leaves of Morus atropurpurea. Chin. Med. 2013, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Pohjala, L.; Tammela, P. Aggregating behavior of phenolic compounds-a source of false bioassay results? Molecules 2012, 17, 10774–10790. [Google Scholar] [CrossRef] [Green Version]
- Ali, H.; Houghton, P.; Soumyanath, A. α-Amylase inhibitory activity of some Malaysian plants used to treat diabetes; with particular reference to Phyllanthus amarus. J. Ethnopharmacol. 2006, 107, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An aggregation advisor for ligand discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, B.Y.; Shelat, A.; Doman, T.N.; Guy, R.K.; Shoichet, B.K. High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol. 2005, 1, 146–148. [Google Scholar] [CrossRef]
- McGovern, S.L.; Helfand, B.T.; Feng, B.; Shoichet, B.K. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003, 46, 4265–4272. [Google Scholar] [CrossRef]
- Szabo, K.; Hamori, C.; Gyemant, G. Gallotannins are non-specific inhibitors of α-amylase: Aggregates are the active species taking part in inhibition. Chem. Biol. Drug Des. 2021, 97, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Wan, C.; Liu, K.; Seeram, N.P. New maplexins F-I and phenolic glycosides from red maple (Acer rubrum) bark. Tetrahedron 2012, 68, 959–964. [Google Scholar] [CrossRef]
- Zaharudin, N.; Salmean, A.A.; Dragsted, L.O. Inhibitory effects of edible seaweeds, polyphenolics and alginates on the activities of porcine pancreatic α-amylase. Food Chem. 2018, 245, 1196–1203. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extracts (200 µg/mL) | α-Glucosidase Assay | α-Amylase Assay | ||
|---|---|---|---|---|
| % Inhibition | IC50 (µg/mL) | % Inhibition | IC50 (µg/mL) | |
| Hexane | 11.67 ± 7.81 | - | NA | - |
| Chloroform | 50.30 ± 1.64 | - | NA | - |
| Ethyl acetate | 50.89 ± 7.34 | - | NA | - |
| Acetone | 96.65 ± 0.26 | 16.61 ± 2.33 | NA | - |
| Methanol | 99.73 ± 0.30 | 6.67 ± 0.30 | 83.84 ± 5.79 | 25.62 ± 4.12 |
| Water | 98.12 ± 0.46 | 13.99 ± 1.34 | NA | - |
| Acarbose | 50.17 ± 1.58 | 214.3 ± 6.23 | 93.22 ± 2.13 | 9.07 ± 0.96 |
| Com. No | RT (min) | UV Max (nm) | Tentative Identification | Molecular Formula | Observed (m/z) | Theoretical (m/z) | Error (ppm) | Fragment Ions-MSn (m/z) | Source/Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.63 | 259 | Sucrose | C12H22O11 | [M + FA]− 387.1135, [M − H]− 341.1078 | [M − H]− 341.1089 | −3.22 | MS2(341): 179 bp, 161, 143, 131, 119, 113, 101 MS3(179): 161, 149, 143 bp, 131, 119, 113, 101, 89, 71 | [19], Kew library |
| 2 | 1.82 | 260 | HHDP-hexoside (isomer 1) | C20H18O14 | [M − H]− 481.0618 | [M − H]− 481.0623 | −1.04 | MS2(481): 321, 301 bp, 300, 275 MS3(301): 284, 257 bp, 229, 201, 185 | [20,21,22] |
| 3 | 2.39 | 262 | HHDP-hexoside (isomer 2) | C20H18O14 | [M − H]− 481.0623 | [M − H]− 481.0623 | 0 | MS2(481): 301 bp, 275 MS3(301): 284, 257 bp, 229, 201, 185 | [20,21,22] |
| 4 | 2.46 | 270 | Gallic acid (isomer 1) | C7H6O5 | [M − H]− 169.0137 | [M − H]− 169.0142 | −2.96 | MS2(169): 151 bp, 125 MS3(151): 123, 95 bp | [23,24] |
| 5 | 2.9 | 270 | Gallic acid (isomer 2) | C7H6O5 | [M − H]− 169.0137 | [M − H]− 169.0142 | −2.96 | MS2(169): 125 bp, 151 MS3(151): 97, 81 bp | [23,24] |
| 6 | 6.47 | 272 | Unknown tannin | >2000 | [M–2H]2− 1067.1188 | >2000 | - | MS2(1067): 377, 885, 933, 977, 1005, 1023 bp MS3(1023): 377, 533, 631, 703, 721, 859, 885, 903 bp, 933, 1005. | - |
| 7 | 7.31 | 272 | Galloyl-HHDP-hexoside | C27H22O18 | [M − H]− 633.0715 | [M − H]− 633.0733 | −2.84 | MS2(633): 463, 301 bp MS3(301): 284, 257 bp, 229, 201,185 | [25] |
| 8 | 7.74 | 273 | Galloyl-bis-HHDP-hexoside (isomer 1) | C41H28O26 | [M − H]− 935.0761 | [M − H]− 935.0796 | −3.74 | MS2(935): 917, 873, 783, 659, 633 bp, 615, 589, 571 MS3(633): 615, 589, 571 bp, 553, 481, 437, 419, 401, 383, 365, 329, 317, 299, 275 | [25,26] |
| 9 | 8.9 | 271 | n.i | - | [M − H]− 603.0678 | - | - | MS2(603): 458 bp MS3(458): 573, 436 bp, 427, 413, 382, 343, 275. | - |
| 10 | 11.5 | 275 | Galloyl-bis-HHDP-hexoside (isomer 2) | C41H28O26 | [M − H]− 935.0757 | [M − H]− 935.0796 | −4.17 | MS2(935): 917, 783, 633 bp, 615, 301 MS3(633): 615, 481, 463, 301 bp | [25,26] |
| 11 | 13.14 | 266 | Myricetin-galloyl-hexoside | C28H24O17 | [M − H]− 631.0913 | [M − H]− 631.0941 | −4.43 | MS2(631): 479 bp, 317 MS3(317): 287, 271, 179, 151 | [27] |
| 12 | 15.03 | 354, 266 | Myricetin-O-rutinoside | C27H30O17 | [M − H]− 625.1390 | [M − H]− 625.1410 | −3.20 | MS2(625): 317, 316 bp, 287, 271 MS3(316): 287, 271, 179, 151 | Fragmentation pattern |
| 13 | 15.37 | 353, 264 | Myricetin-O-hexoside | C21H20O13 | [M − H]− 479.0824 | [M − H]− 479.0831 | −1.46 | MS2(479): 317, 316 bp MS3(316): 287 bp, 271, 179, 151 | [27] |
| 14 | 17.27 | 361 | Ellagic acid-O-pentoside | C19H14O12 | [M − H]− 433.0401 | [M − H]− 433.0413 | −2.77 | MS2(433): 301 bp, 300 MS3(301): 284, 257 bp, 244, 229, 201, 185 | [28,29], Kew library |
| 15 | 17.78 | 361 | Ellagic acid-O-rhamnoside | C20H16O12 | [M − H]− 447.0559 | [M − H]− 447.0569 | −2.24 | MS2(447): 301 bp, 300 MS3(301): 284, 271, 257 bp, 244, 229, 201, 185 | [28,29], Kew library |
| 16 | 18.74 | 355 | Quercetin-O-rutinoside | C27H30O16 | [M − H]− 609.1437 | [M − H]− 609.1461 | −3.94 | MS2(609): 301 bp, 300, 271, 255 MS3(301): 271, 255, 179 bp, 151 | [30] |
| 17 | 19.19 | 367 | Ellagic acid | C14H6O8 | [M − H]− 300.9984 | [M − H]− 300.9990 | −1.99 | MS2(301): 284, 257 bp, 244, 229, 201, 185 | [28,29], Kew library |
| 18 | 19.66 | 359 | Methoxymyricetin-O-rutinoside | C28H32O17 | [M − H]− 639.1545 | [M − H]− 639.1567 | −3.44 | MS2(639): 607, 331 bp, 316, 315, 287, 271 MS3(316): 287 bp, 271, 259, 243, 178, 151 | Fragmentation pattern |
| 19 | 20.96 | 348 | Kaempferol-O-rutinoside | C27H30O15 | [M − H]− 593.1491 | [M − H]− 593.1512 | −3.54 | MS2(593): 327, 285 bp, 255, 227 MS3(285): 267, 257, 255 bp, 241, 229, 227, 213, 197, 151 | [30,31,32] |
| 20 | 22.48 | 355 | Methoxyquercetin-O-rutinoside (isomer 1) | C28H32O16 | [M − H]− 623.1597 | [M − H]− 623.1618 | −3.37 | MS2(623): 357, 315 bp, 300, 271 MS3(315): 300 bp, 299 MS3(300): 271 bp, 255 | [18,30,33] |
| 21 | 22.88 | 355 | Methoxyquercetin-O-rutinoside (isomer 2) | C28H32O16 | [M − H]− 623.1592 | [M − H]− 623.1618 | −4.17 | MS2(623): 357, 315 bp, 300, 271 MS3(315): 300 bp, 299 MS3(300): 271 bp, 255 | [18,30,33] |
| 22 | 23.20 | 359 | Dimethoxymyricetin-O-rutinoside | C29H34O17 | [M − H]− 653.1700 | [M − H]− 653.1723 | −3.52 | MS2(653): 345 bp, 330, 315, 287 MS3(345): 330 bp, 315 MS3(315): 287 bp, 271, 259, 243 | [18] |
| 23 | 26.92 | 311, 265 | Quercetin-O-caffeoyl-rhamnoside | C30H26O14 | [M − H]− 609.1229 | [M − H]− 609.1250 | −3.45 | MS2(609): 463 bp, 301 MS3(301): 271 bp, 255, 179, 151 | Fragmentation Pattern |
| 24 | 27.11 | 311, 267 | Methoxymyricetin-O-caffeoyl-rhamnoside | C31H28O15 | [M − H]− 639.1338 | [M − H]− 639.1355 | −2.66 | MS2(639): 493 bp, 331 MS3(331): 316 bp, 315, 179, 151 | Fragmentation Pattern |
| 25 | 27.64 | 281, 265 | n.i. | - | [M + FA]− 711.3938, [M − H]− 665.3620 | - | - | MS2(711): 665, 503 bp MS3(503): 505, 485, 453, 441 bp, 421, 409, 403 | - |
| 26 | 30.08 | 311, 266 | Dimethoxymyricetin-O-caffeoyl-rhamnoside | C32H30O15 | [M − H]− 653.1491 | [M − H]− 653.1512 | −3.32 | MS2(653): 345 bp, 330, 315, 287 MS3(345): 330 bp, 315 MS3(315): 287 bp, 271, 259, 243 | Fragmentation Pattern |
| 27 | 30.38 | 315, 265 | Methoxyquercetin-O-caffeoyl-rhamnoside | C31H28O14 | [M − H]− 623.1386 | [M − H]− 623.1406 | −3.21 | MS2(623): 315 bp, 300 MS3(300): 271 bp, 255, 227, 151 | Fragmentation Pattern |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khojah, A.A.; Padilla-González, G.F.; Bader, A.; Simmonds, M.J.S.; Munday, M.; Heinrich, M. Barbeya oleoides Leaves Extracts: In Vitro Carbohydrate Digestive Enzymes Inhibition and Phytochemical Characterization. Molecules 2021, 26, 6229. https://doi.org/10.3390/molecules26206229
Khojah AA, Padilla-González GF, Bader A, Simmonds MJS, Munday M, Heinrich M. Barbeya oleoides Leaves Extracts: In Vitro Carbohydrate Digestive Enzymes Inhibition and Phytochemical Characterization. Molecules. 2021; 26(20):6229. https://doi.org/10.3390/molecules26206229
Chicago/Turabian StyleKhojah, Alaa A., Guillermo F. Padilla-González, Ammar Bader, Monique J. S. Simmonds, Michael Munday, and Michael Heinrich. 2021. "Barbeya oleoides Leaves Extracts: In Vitro Carbohydrate Digestive Enzymes Inhibition and Phytochemical Characterization" Molecules 26, no. 20: 6229. https://doi.org/10.3390/molecules26206229
APA StyleKhojah, A. A., Padilla-González, G. F., Bader, A., Simmonds, M. J. S., Munday, M., & Heinrich, M. (2021). Barbeya oleoides Leaves Extracts: In Vitro Carbohydrate Digestive Enzymes Inhibition and Phytochemical Characterization. Molecules, 26(20), 6229. https://doi.org/10.3390/molecules26206229