
Analysis of the Structure-Function-Dynamics Relationships of GALT Enzyme and of Its Pathogenic Mutant p.Q188R: A Molecular Dynamics Simulation Study in Different Experimental Conditions

 , ,
, , 
Abstract
:1. Introduction
2. Results and Discussion
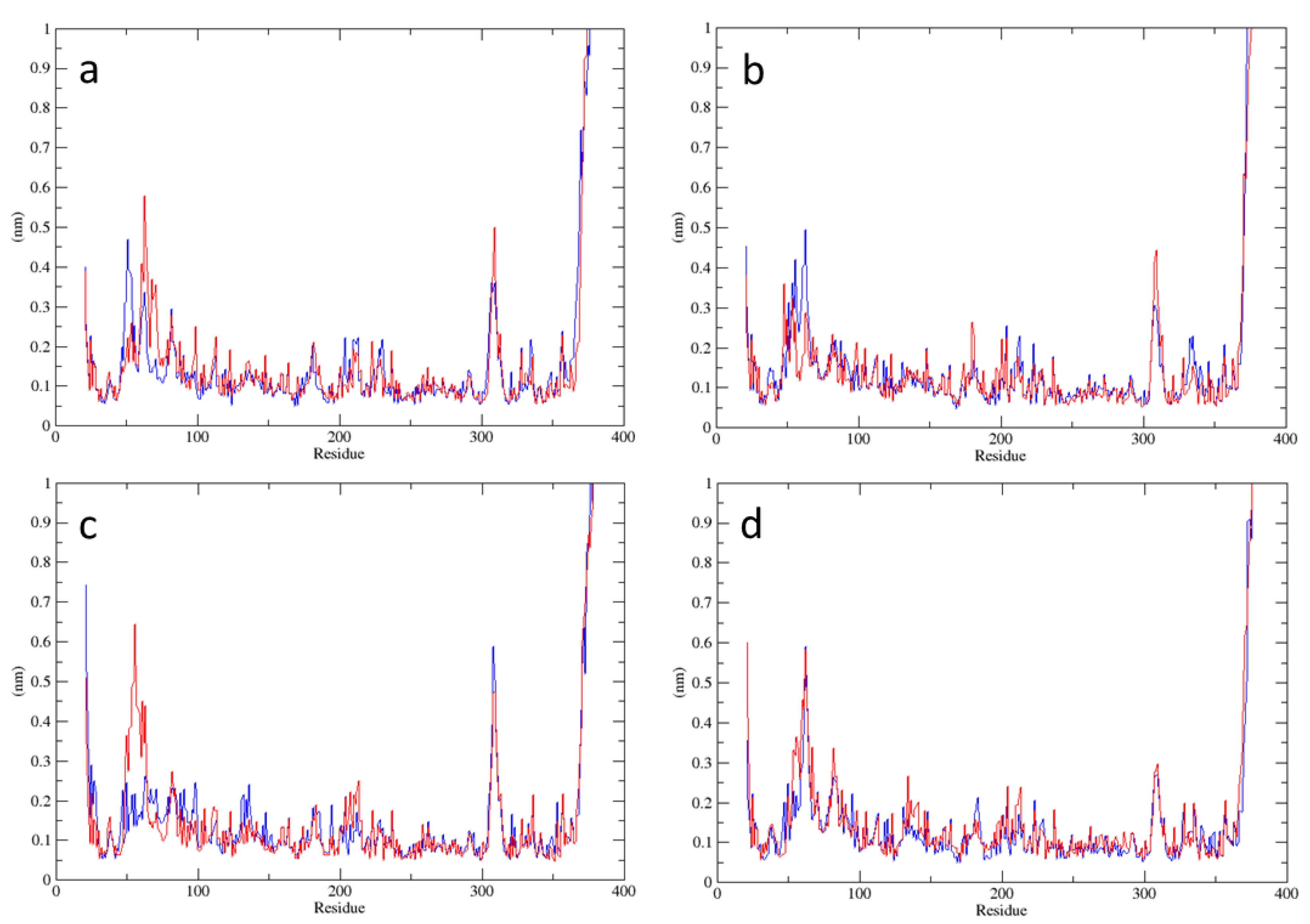
2.1. Analysis of MD Simulations at 310 K
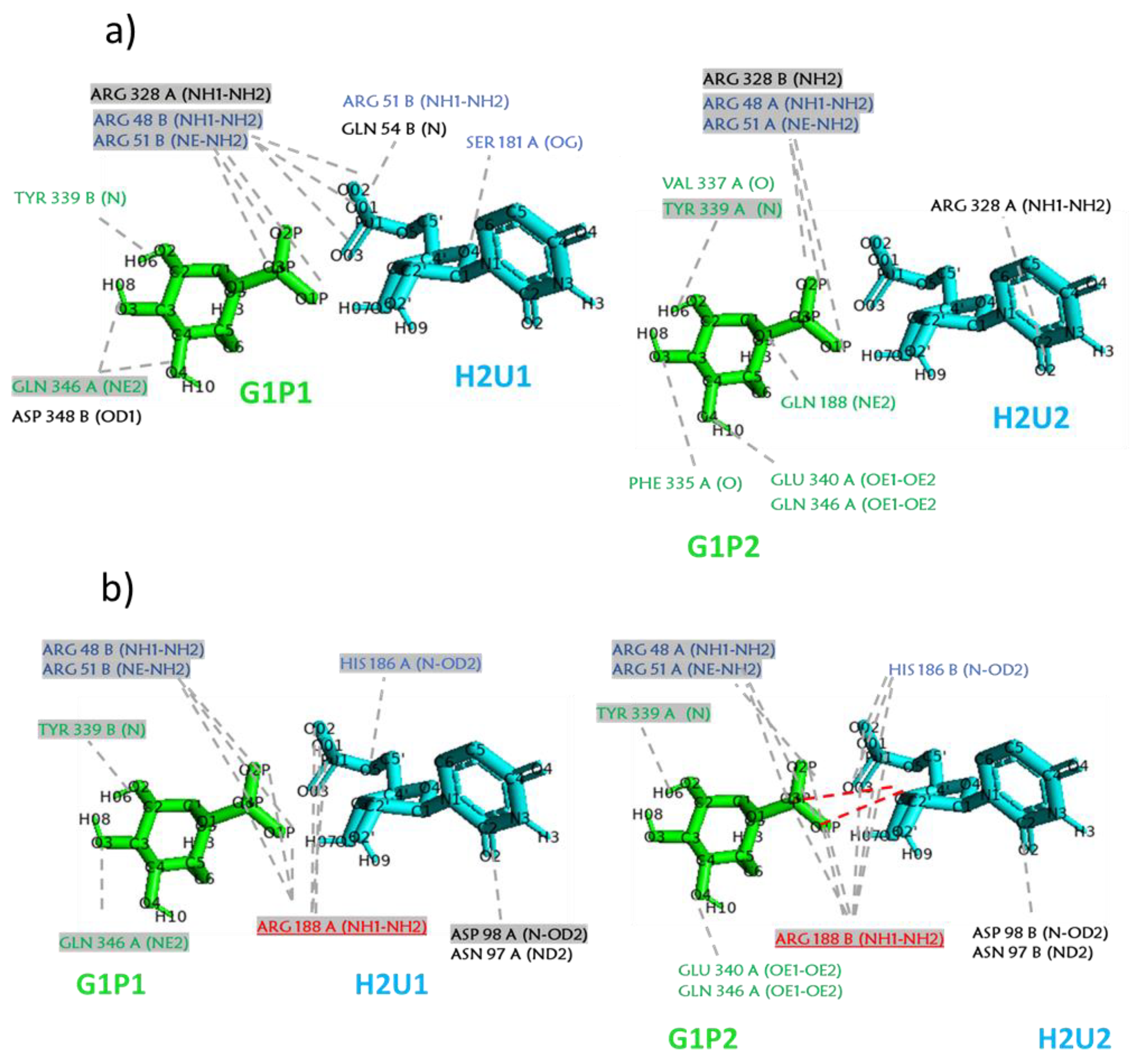
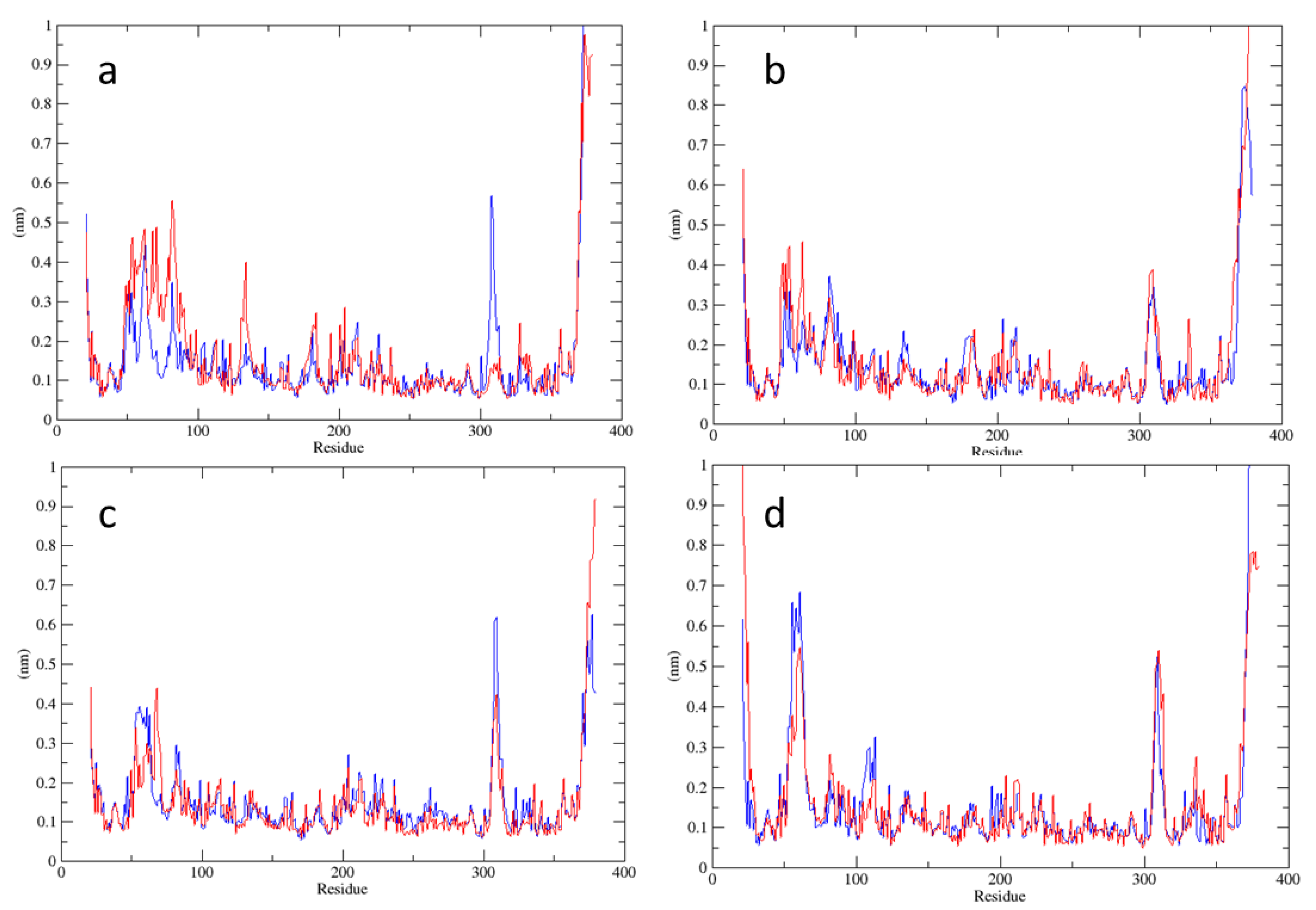
2.2. Analysis of MD Simulations at 334 K
3. Materials and Methods
3.1. Starting Structures
3.2. MD Simulations and Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Acosta, P.B.; Gross, K.C. Hidden sources of galactose in the environment. Eur. J. Pediatr. 1995, 154, S87–S92. [Google Scholar] [CrossRef]
- Leloir, L.F. Two decades of research on the biosynthesis of saccharides. Science 1971, 172, 1299–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, H.M.; Rayment, I.; Thoden, J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol Chem. 2003, 278, 43885–43888. [Google Scholar] [CrossRef] [Green Version]
- Berry, G.T. Classic galactosemia and clinical variant galactosemia. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2020; 4 February 2000 [updated 2 July 2020]. [Google Scholar]
- Marabotti, A.; Facchiano, A.M. Homology modeling studies on human galactose-1-phosphate uridylyltransferase and on its galactosemia-related mutant Q188R provide an explanation of molecular effects of the mutation on homo- and heterodimers. J. Med. Chem. 2005, 48, 773–779. [Google Scholar] [CrossRef]
- Facchiano, A.; Marabotti, A. Analysis of galactosemia-linked mutations of GALT enzyme using a computational biology approach. Protein Eng. Des. Sel. 2010, 23, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Acierno, A.; Facchiano, A.; Marabotti, A. GALT protein database, a bioinformatics resource for the management and analysis of structural features of a galactosemia-related protein and its mutants. Genom. Proteom. Bioinform. 2009, 7, 71–76. [Google Scholar] [CrossRef] [Green Version]
- d’Acierno, A.; Facchiano, A.; Marabotti, A. GALT protein database: Querying structural and functional features of GALT enzyme. Hum. Mutat. 2014, 35, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- d’Acierno, A.; Scafuri, B.; Facchiano, A.; Marabotti, A. The evolution of a Web resource: The Galactosemia Proteins Database 2.0. Hum. Mutat. 2018, 39, 52–60. [Google Scholar] [CrossRef]
- McCorvie, T.J.; Kopec, J.; Pey, A.L.; Fitzpatrick, F.; Patel, D.; Chalk, R.; Shrestha, L.; Yue, W.W. Molecular basis of classic galactosemia from the structure of human galactose 1-phosphate uridylyltransferase. Hum. Mol. Genet. 2016, 25, 2234–2244. [Google Scholar] [CrossRef]
- d’Acierno, A.; Facchiano, A.; Brando, F.; Scafuri, B.; Marabotti, A. Expansion of Galactosemia Proteins Database 2.0 with a new galactosemia related protein [version 1; not peer reviewed]. F1000Research 2020, 9, 499. [Google Scholar] [CrossRef]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Moroni, E.; Agard, D.A.; Colombo, G. The structural asymmetry of mitochondrial Hsp90 (Trap1) determines fine tuning of functional dynamics. J. Chem Theory Comput. 2018, 14, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, C.; Moroni, E.; Ferraro, M.; Laquatra, C.; Cannino, G.; Masgras, I.; Negro, A.; Quadrelli, P.; Rasola, A.; Colombo, G. Rational design of allosteric and selective inhibitors of the molecular chaperone TRAP1. Cell Rep. 2020, 31, 107531. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Facchiano, A.; Rachamadugu, R.; Calderon, F.; Mao, R.; Milanesi, L.; Marabotti, A.; Lai, K. Correlation assessment among clinical phenotypes, expression analysis and molecular modeling of 14 novel variations in the human galactose-1-phosphate uridylyltransferase gene. Hum. Mutat. 2012, 33, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringe, D.; Petsko, G.A. What are pharmacological chaperones and why are there interesting? J. Biol. 2009, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timson, D.J. The molecular basis of galactosemia—Past, present and future. Gene 2016, 589, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdino, A.; D’Urso, G.; Tammone, C.; Scafuri, B.; Catapano, L.; Marabotti, A. Study of the interactions of Arginine with wild-type GALT enzyme and the classic galactosemia-related mutant p.Q188R by a computational approach. Molecules. (in press).
- Coelho, A.I.; Trabuco, M.; Silva, M.J.; de Almeida, I.T.; Leandro, P.; Rivera, I.; Vicente, J.B. Arginine functionally improves clinically relevant human galactose-1-phosphate uridylyltransferase (GALT) variants expressed in a prokaryotic model. JIMD Rep. 2015, 23, 1–6. [Google Scholar] [PubMed] [Green Version]
- Coelho, A.I.; Trabuco, M.; Ramos, R.; Silva, M.J.; Tavares de Almeida, I.; Leandro, P.; Rivera, I.; Vicente, J.B. Functional and structural impact of the most prevalent missense mutations in classic galactosemia. Mol. Genet. Genom. Med. 2014, 2, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Castrignanò, T.; Gioiosa, S.; Flati, T.; Cestari, M.; Picardi, E.; Chiara, M.; Fratelli, M.; Amente, S.; Cirilli, M.; Tangaro, M.A.; et al. ELIXIR-IT HPC@CINECA: High performance computing resources for the bioinformatics community. BMC Bioinform. 2020, 21, 352. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Prot Chem. 2003, 66, 27–85. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, J.L.F.; Vega, C. A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem Phys. 2005, 123, 234505. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Berendsen, H.J.C. A leap-frog algorithm for stochastic dynamics. Mol. Simul. 1988, 1, 173–185. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Verlet, L. Computer “experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
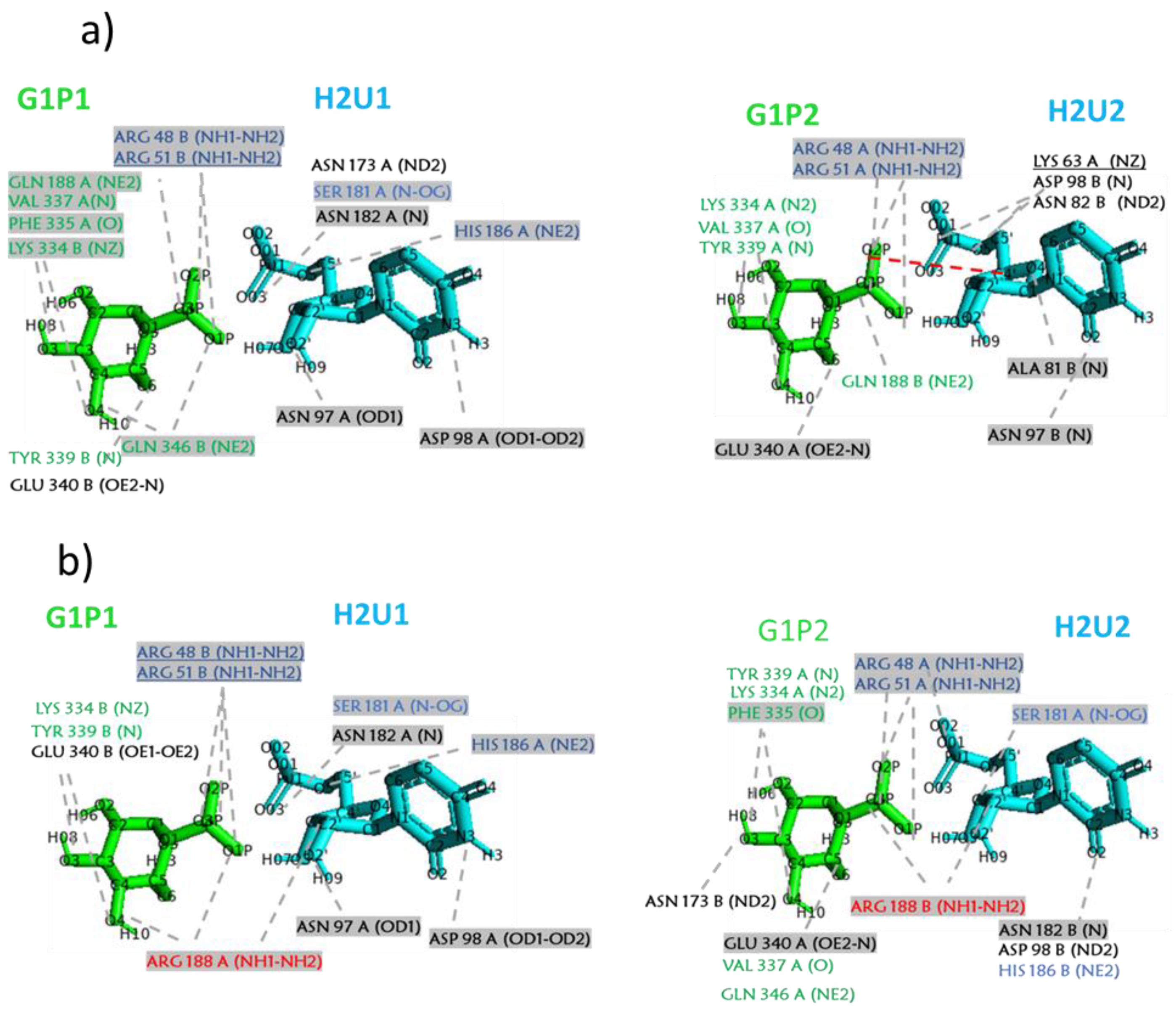{kind=link}
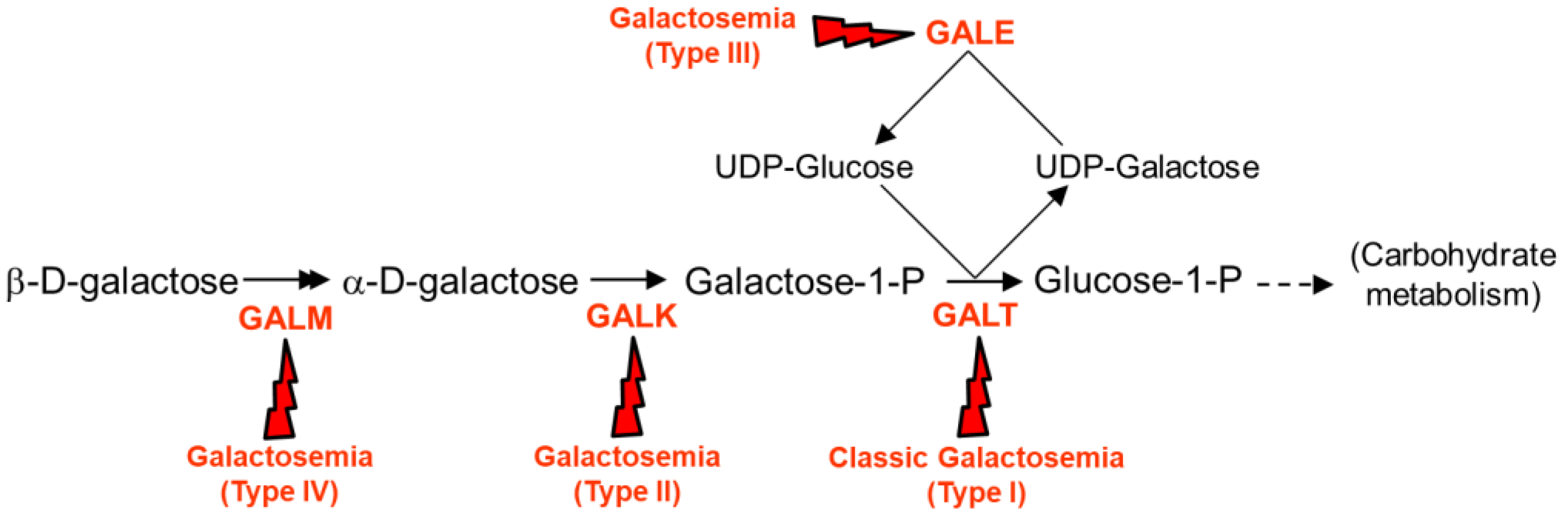{kind=link}
| wtGALT | p.Gln188Arg | wtGALT + Ligands | p.Gln188Arg + Ligands |
|---|---|---|---|
| Average Number of H-Bonds per Timeframe: 27 | Average Number of H-Bonds per Timeframe: 29 | Average Number of H-Bonds per Timeframe: 30 | Average Number of H-Bonds per Timeframe: 25 |
| ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B |
| ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A |
| TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B |
| TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A |
| ILE198A-ALA343B | ILE198A-ALA343B | ARG48A-PHE99B | ARG48A-PHE99B |
| ILE198B-ALA343A | ARG48A-PRO100 | ILE198A-ALA343B | ASP197A-GLN344B |
| HIS301A-LEU342B | HIS301A-LEU342B | ILE198B-ALA343A | SER45A-ALA101B |
| ASP197A-GLN334B | ASP197A-GLN334B | HIS47B-PRO100A | ILE198A-ALA343B |
| ARG48A-PHE99B | GLN30A-GLN103B | ASP197B-GLN344A | HIS47B-PRO100A |
| SER45A-ALA101B | GLN30A-ALA122B | TRP41A-ASP197B | TRP41A-ASP197B |
| GLN30A-GLN103B | TRP41A-ASP197B | TRP41B-ASP197A | TRP41B-ASP197A |
| GLN30A-ALA122B | HIS47A-PRO100B | GLN30B-GLN103A | GLN30B-GLN103A |
| TRP41A-ASP197B | ARG228B-ASP113A | GLN30B-ALA122A | GLN30B-ALA122A |
| HIS47A-PRO100B | ARG333B-GLU58A | ARG228A-ASP113B | ARG228A-ASP113B |
| ARG228B-ASP113A | ARG228A-ASP113B | ARG228B-ASP113A | ARG228B-ASP113A |
| ARG333B-GLU58A | ARG201A-ASP39B | GLY338A-SER297B | GLY338A-SER297B |
| SER45B-ALA101A | GLN103A-GLN30B | SER45B-ALA101A | SER45B-ALA101A |
| ARG51B-ASP98A | GLY338B-SER297A | GLY338B-SER297A | GLN224A-HIS114B |
| ARG51B-ASP98A | ARG48B-PHE99A | ||
| GLN30A-GLN103B | |||
| GLN30A-ALA122B | |||
| ARG48B-PHE99A |
| wtGALT | p.Gln188Arg | wtGALT + Ligands | p.Gln188Arg + Ligands |
|---|---|---|---|
| GLU58A-ARG333B | GLU58A-ARG333B | ||
| ASP113B-ARG228A | ASP113B-ARG228A | ASP113B-ARG228A | |
| ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B |
| ASP98A-ARG51B | ASP98A-ARG51B | ASP98A-ARG51B |
| wtGALT | p.Gln188Arg | wtGALT + Ligands | p.Gln188Arg + Ligands |
|---|---|---|---|
| Average Number of H-Bonds per Timeframe: 27 | Average Number of H-Bonds per Timeframe: 28 | Average Number of H-Bonds per Timeframe: 27 | Average Number of H-Bonds per Timeframe: 26 |
| ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B | ILE32A-LYS120B |
| ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A | ILE32B-LYS120A |
| TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B | TYR34A-GLN118B |
| ASP197A-GLN344B | TYR34B-GLN118A | TYR34B-GLN118A | TYR34B-GLN118A |
| ILE198A-ALA343B | ASP197A-GLN344B | ARG48A-PHE99B | ARG48A-PHE99B |
| ARG48A-PHE99B | ILE198A-ALA343B | SER45B-ALA101A | SER45A-ALA101B |
| SER45A-ALA101B | GLN30A-GLN103B | ASP197A-GLN344B | ASP197B-GLN344A |
| HIS47B-PRO100A | GLN30A-ALA122B | ILE198A-ALA343B | ILE198B-ALA343A |
| HIS301B-LEU342A | ARG228A-ASP113B | HIS301A-LEU342B | HIS301B-LEU342A |
| TRP41B-ASP197A | ARG228B-ASP113A | GLY338A-SER297B | GLY338A-SER297B |
| GLY338B-SER297A | TRP41B-ASP197A | GLY338B-SER297A | GLY338B-SER297A |
| ARG333B-GLU58A | GLY338B-SER297A | ARG48B-PHE99A | ARG228A-ASP113B |
| GLN103B-GLN30A | ARG333A-GLU58B | GLN30B-GLN103A | ARG228B-ASP113A |
| GLN30A-ALA122B | SER297B-VAL337A | GLN30B-ALA122B | ARG48B-PHE99A |
| ARG228B-ASP113A | TRP41A-ASP197B | TRP41A-ASP197B | TRP41A-ASP197B |
| ARG228A-ASP113B | GLN169B-LEU342A | TRP41B-ASP197A | TRP41B-ASP197A |
| GLN30B-GLN103A | SER45B-ALA101A | ARG228A-ASP113B | ALA122A-GLN30B |
| GLN30B-ALA122A | GLN56A-PRO59B | ARG228B-ASP113A | |
| ARG201B-ASP39A | ARG48B-GLN103A | ||
| HIS47A-PRO100B | |||
| GLY338A-SER297B |
| wtGALT | p.Gln188Arg | wtGALT + Ligands | p.Gln188Arg + Ligands |
|---|---|---|---|
| GLU58A-ARG333B | GLU58A-ARG333B | ||
| ASP113B-ARG228A | ASP113B-ARG228A | ASP113B-ARG228A | ASP113B-ARG228A |
| ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B | ASP113A-ARG228B |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdino, A.; D’Urso, G.; Tammone, C.; Scafuri, B.; Marabotti, A. Analysis of the Structure-Function-Dynamics Relationships of GALT Enzyme and of Its Pathogenic Mutant p.Q188R: A Molecular Dynamics Simulation Study in Different Experimental Conditions. Molecules 2021, 26, 5941. https://doi.org/10.3390/molecules26195941
Verdino A, D’Urso G, Tammone C, Scafuri B, Marabotti A. Analysis of the Structure-Function-Dynamics Relationships of GALT Enzyme and of Its Pathogenic Mutant p.Q188R: A Molecular Dynamics Simulation Study in Different Experimental Conditions. Molecules. 2021; 26(19):5941. https://doi.org/10.3390/molecules26195941
Chicago/Turabian StyleVerdino, Anna, Gaetano D’Urso, Carmen Tammone, Bernardina Scafuri, and Anna Marabotti. 2021. "Analysis of the Structure-Function-Dynamics Relationships of GALT Enzyme and of Its Pathogenic Mutant p.Q188R: A Molecular Dynamics Simulation Study in Different Experimental Conditions" Molecules 26, no. 19: 5941. https://doi.org/10.3390/molecules26195941
APA StyleVerdino, A., D’Urso, G., Tammone, C., Scafuri, B., & Marabotti, A. (2021). Analysis of the Structure-Function-Dynamics Relationships of GALT Enzyme and of Its Pathogenic Mutant p.Q188R: A Molecular Dynamics Simulation Study in Different Experimental Conditions. Molecules, 26(19), 5941. https://doi.org/10.3390/molecules26195941