
Multi-Targeted Molecular Docking, Pharmacokinetics, and Drug-Likeness Evaluation of Okra-Derived Ligand Abscisic Acid Targeting Signaling Proteins Involved in the Development of Diabetes

 ,
,  ,
,  ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results & Discussion
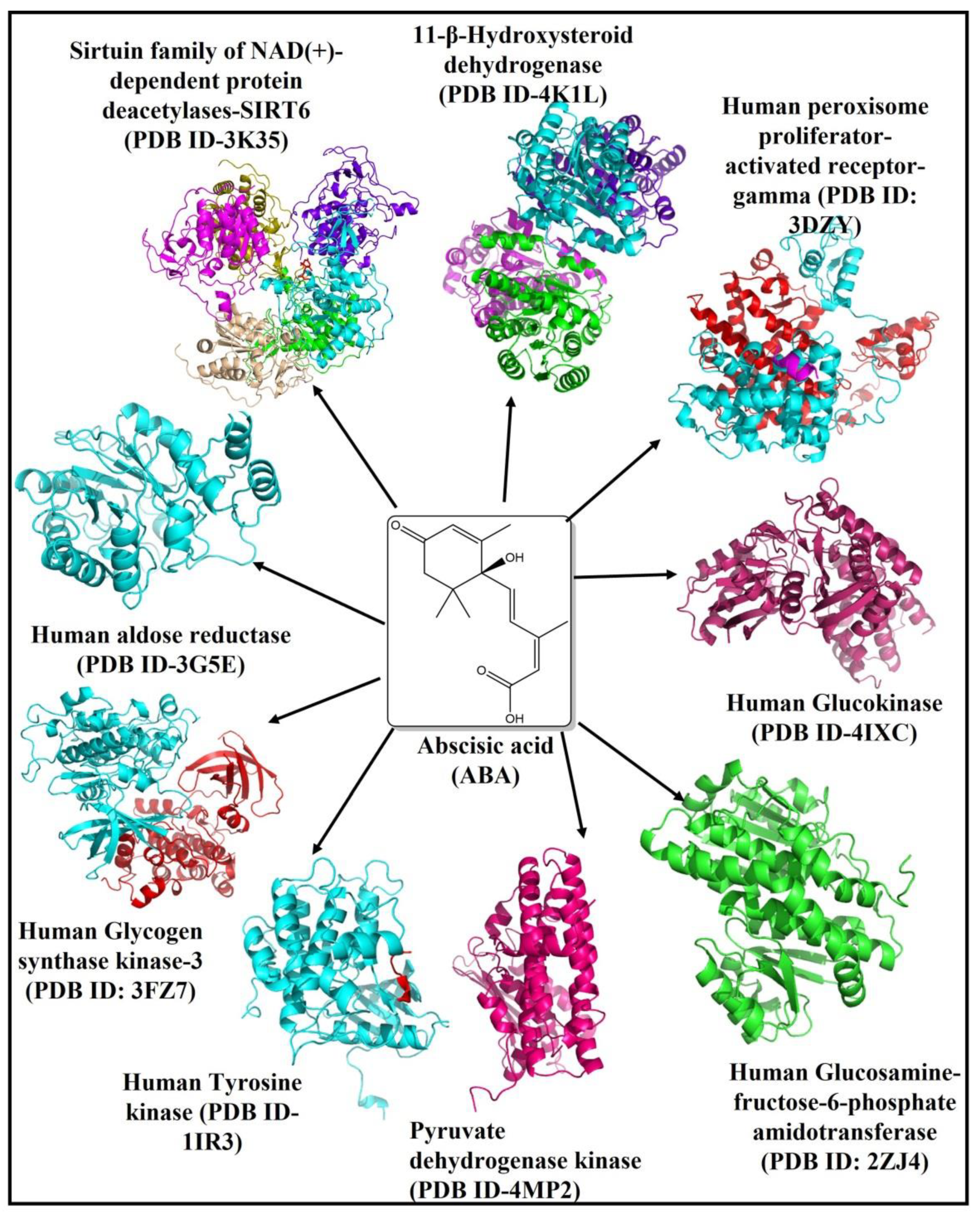
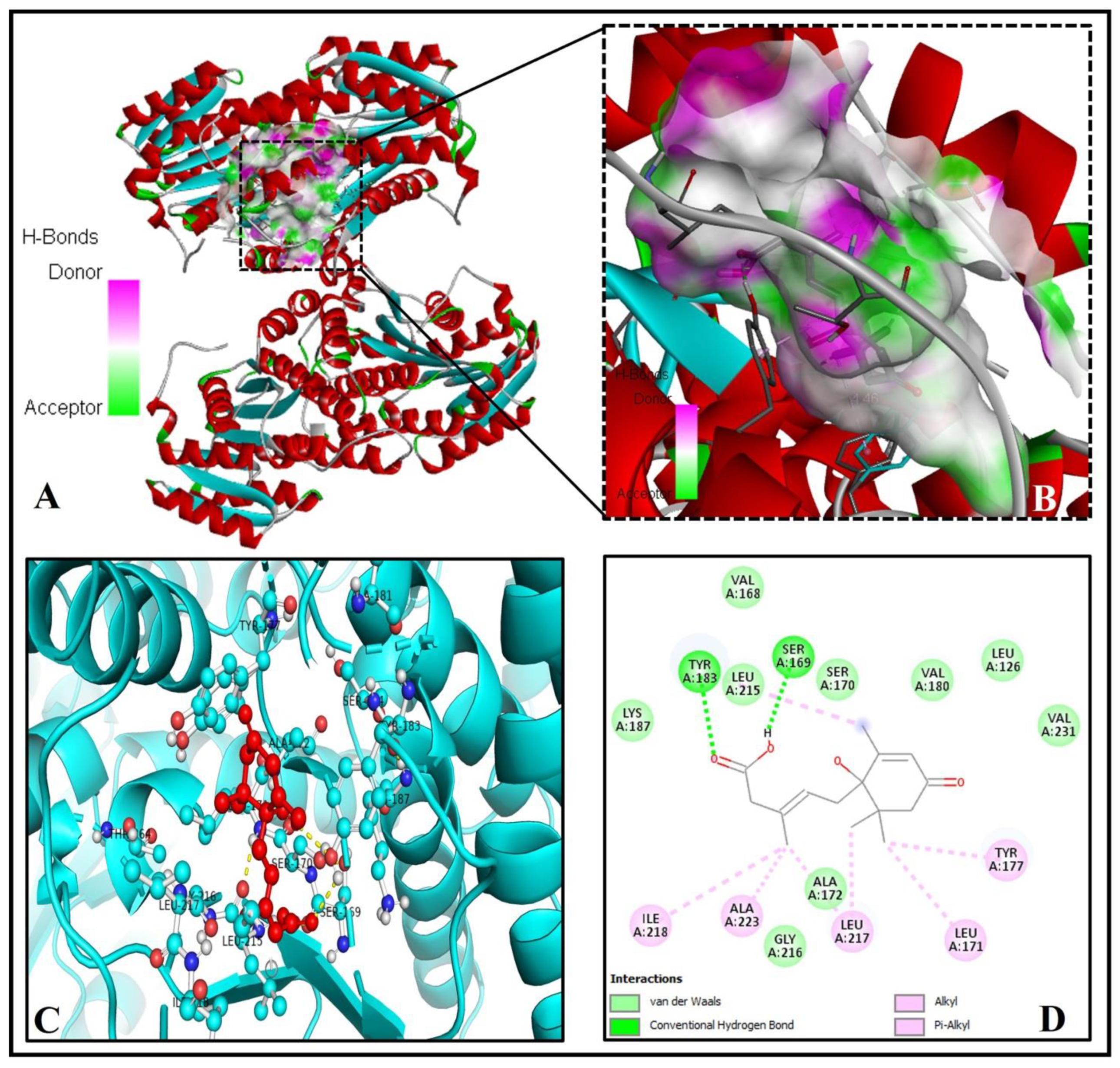
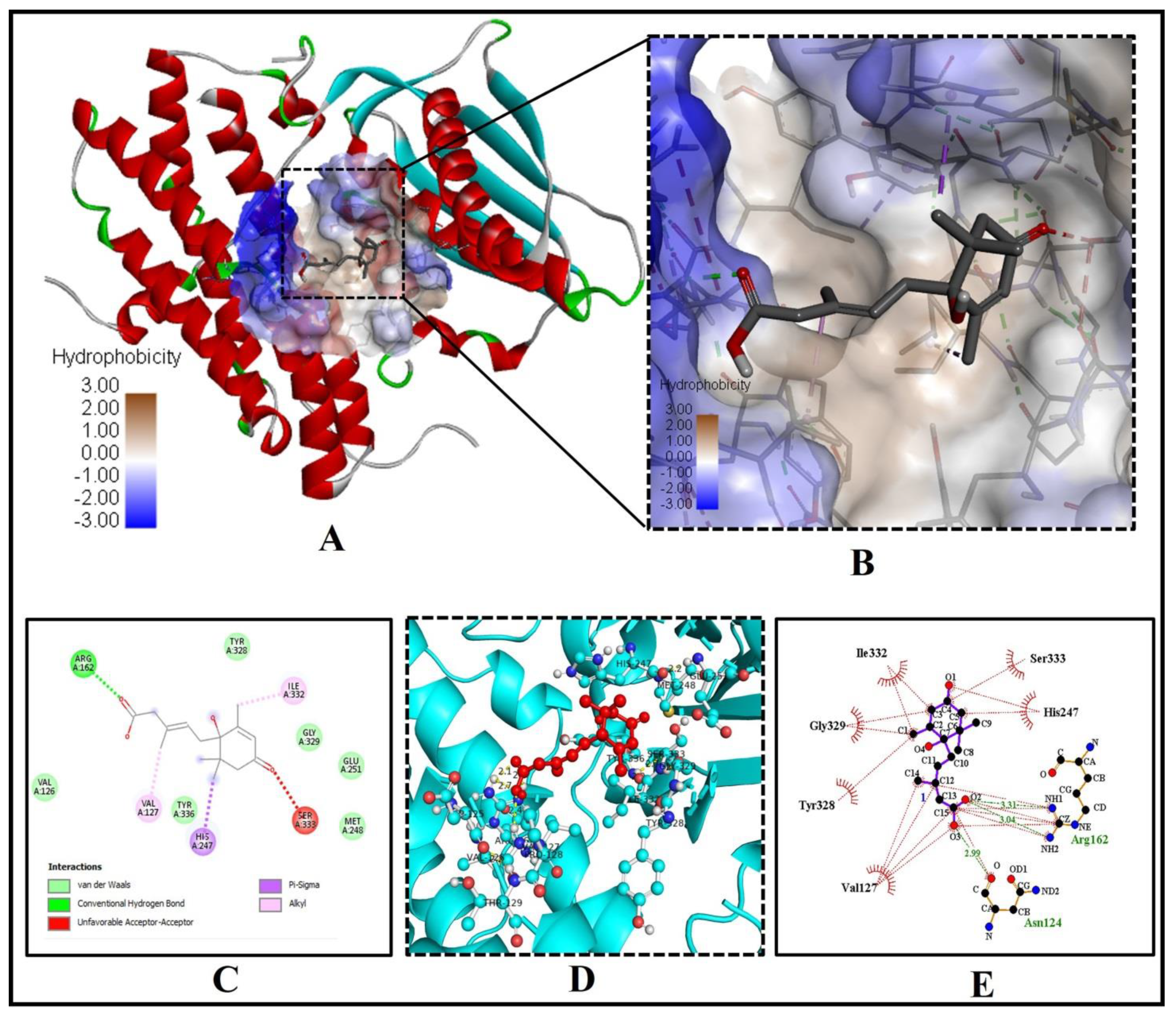
2.1. Abscisic Acid Is a Potent Inhibitor of the Human 11β-Hydroxysteroid Dehydrogenase Type 1 (11β-HSD1) Enzyme
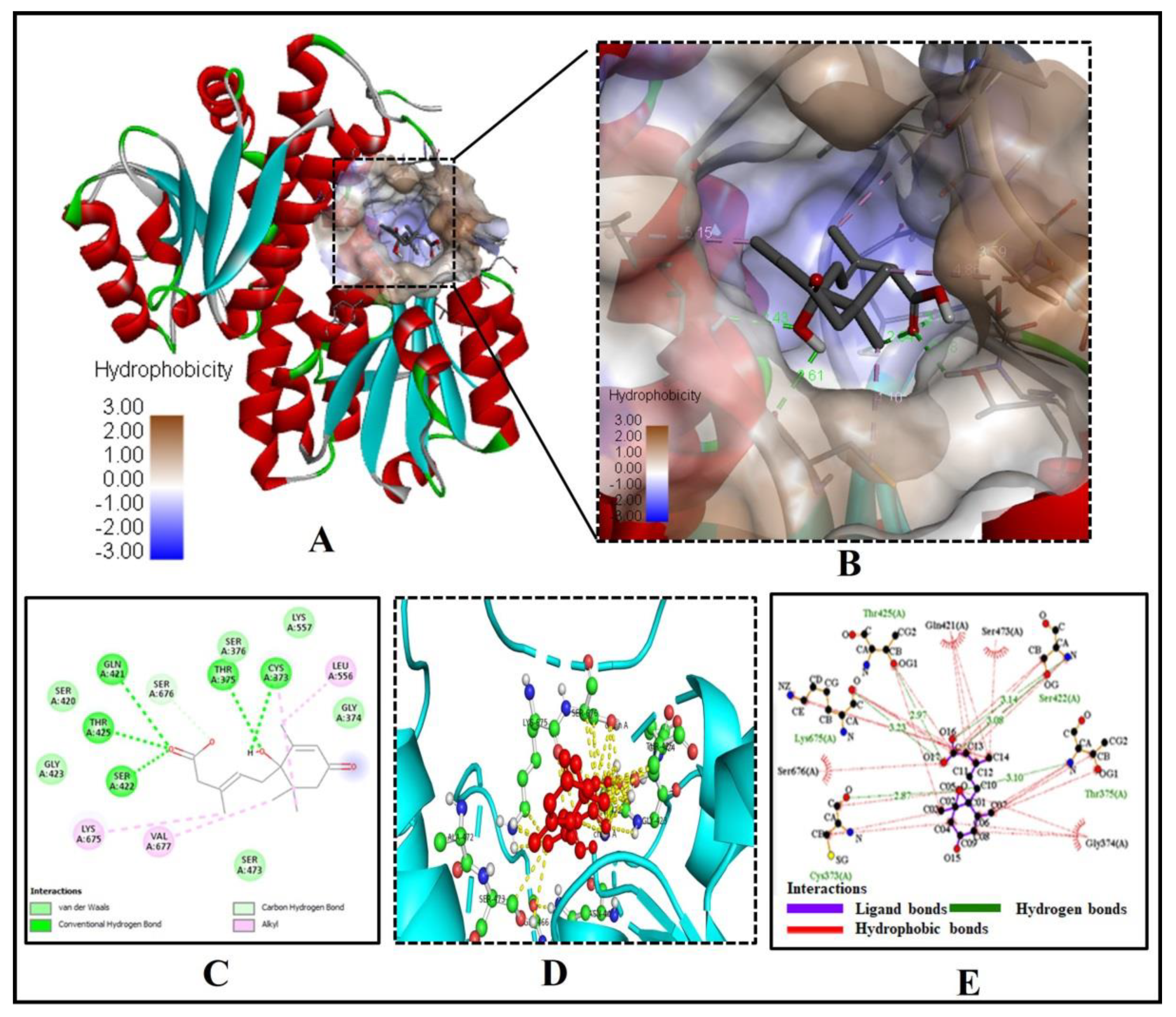
2.2. Abscisic Acid Significantly Binds and Inhibits Glutamine: Fructose-6-Phosphate Amidotransferase (GFAT) Effectively
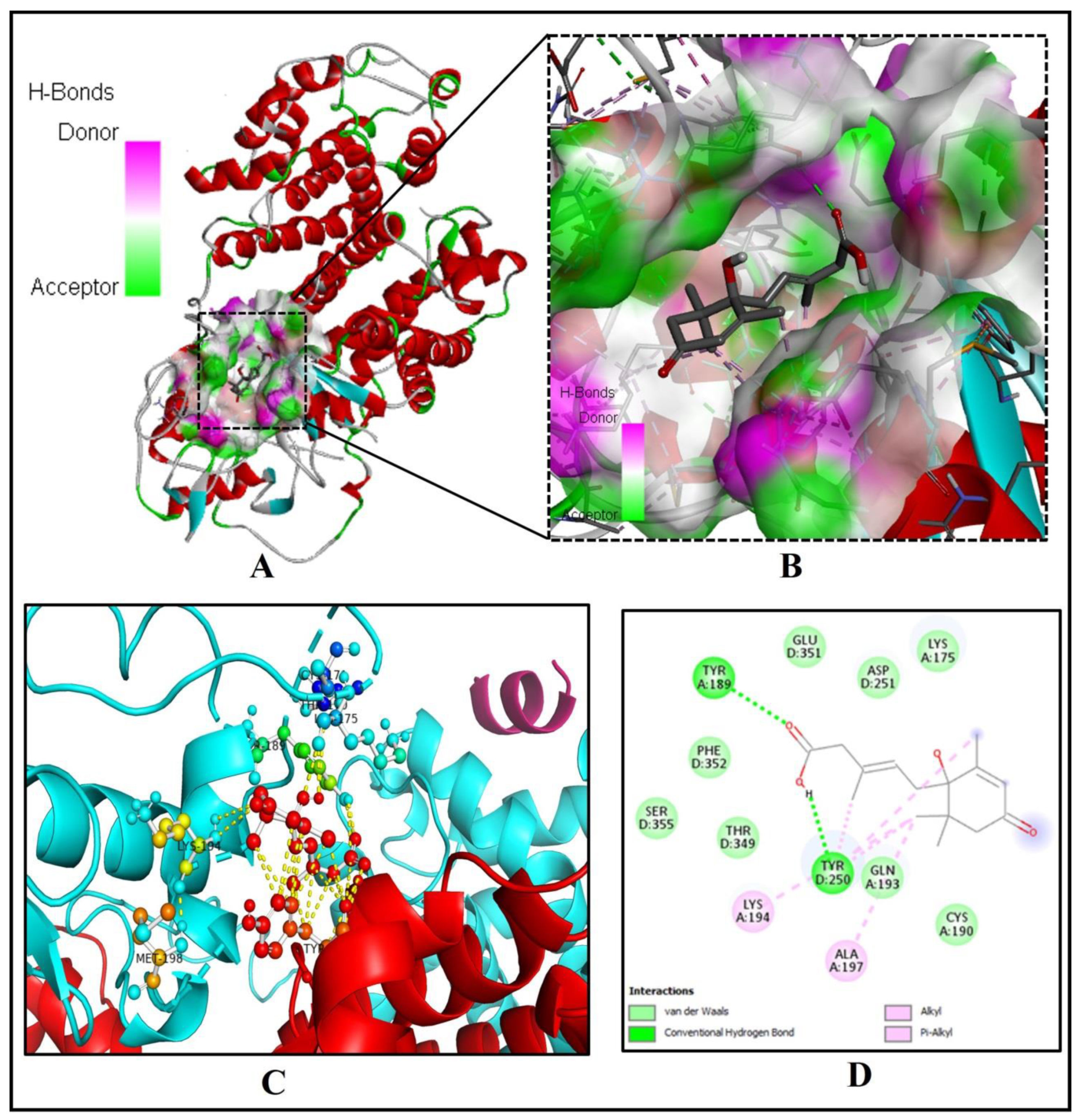
2.3. Binding of Abscisic Acid to the Catalytic Pocket of the Human Peroxisome Proliferator-Activated Receptor-Gamma (PPAR-Gamma)
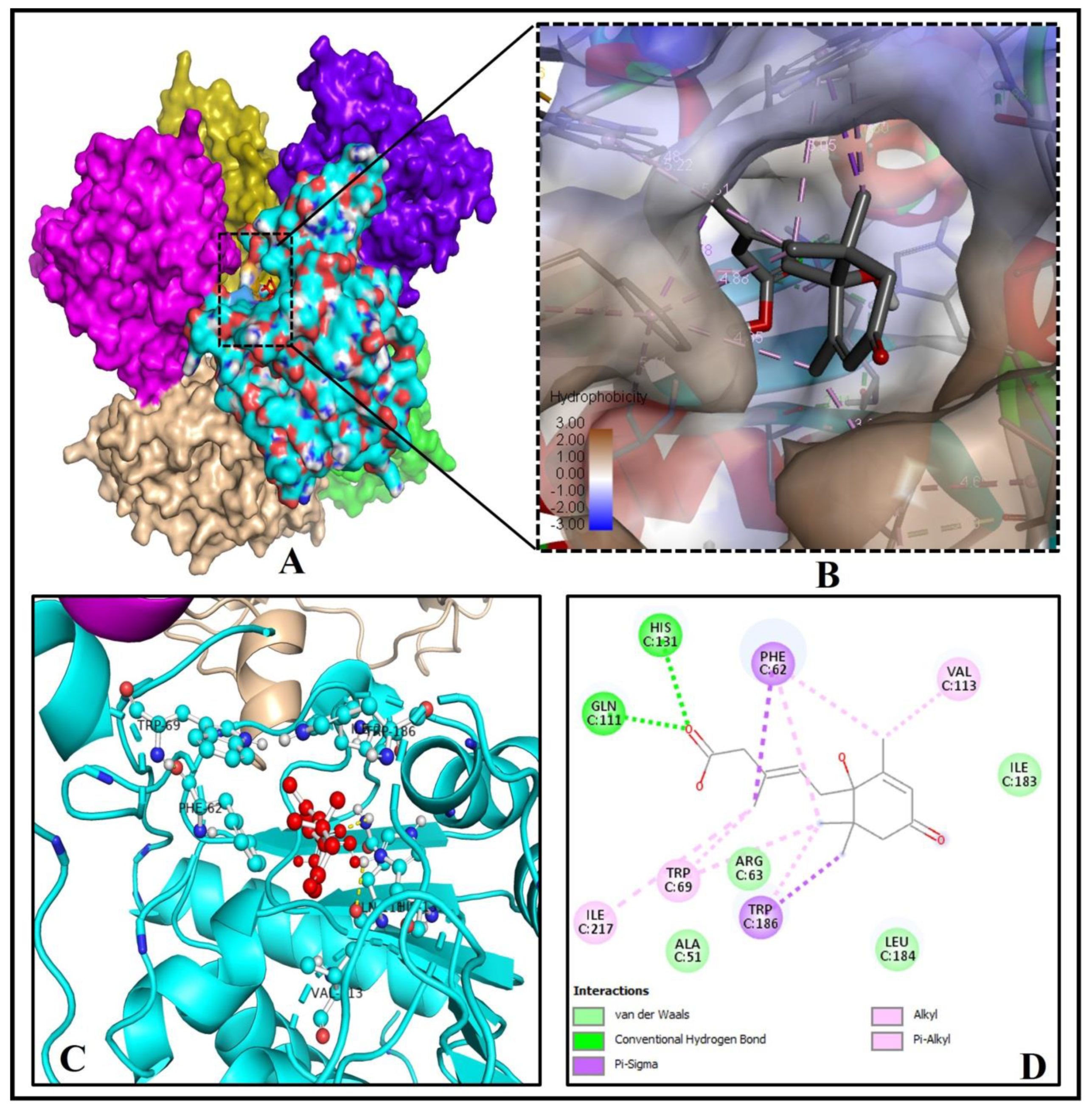
2.4. The Binding Pattern of ABA with Human Mono-ADP-Ribosyl Transferase Sirtuin-6 (SIRT6)
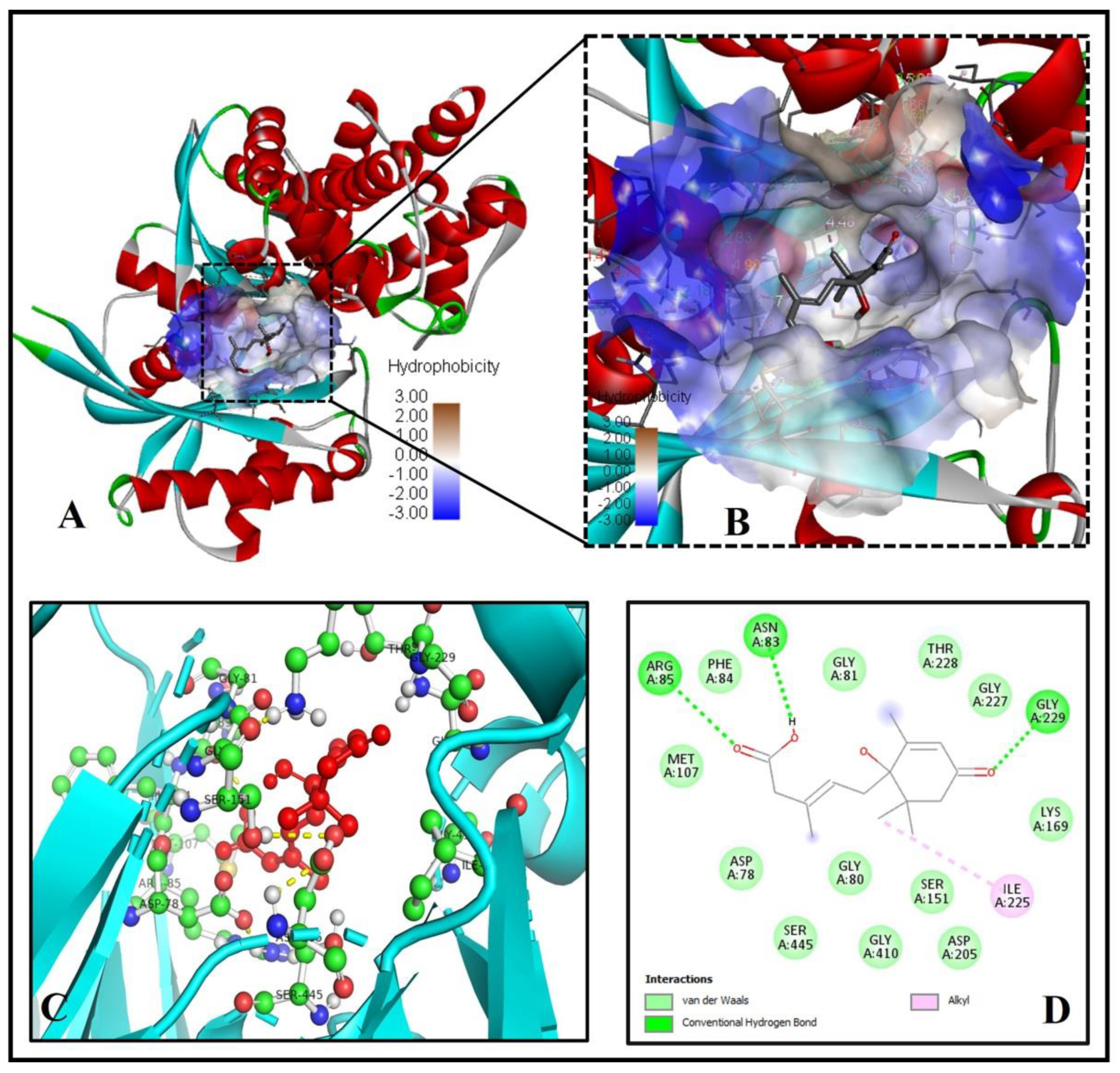
2.5. The Binding Pattern of ABA with Glucokinase
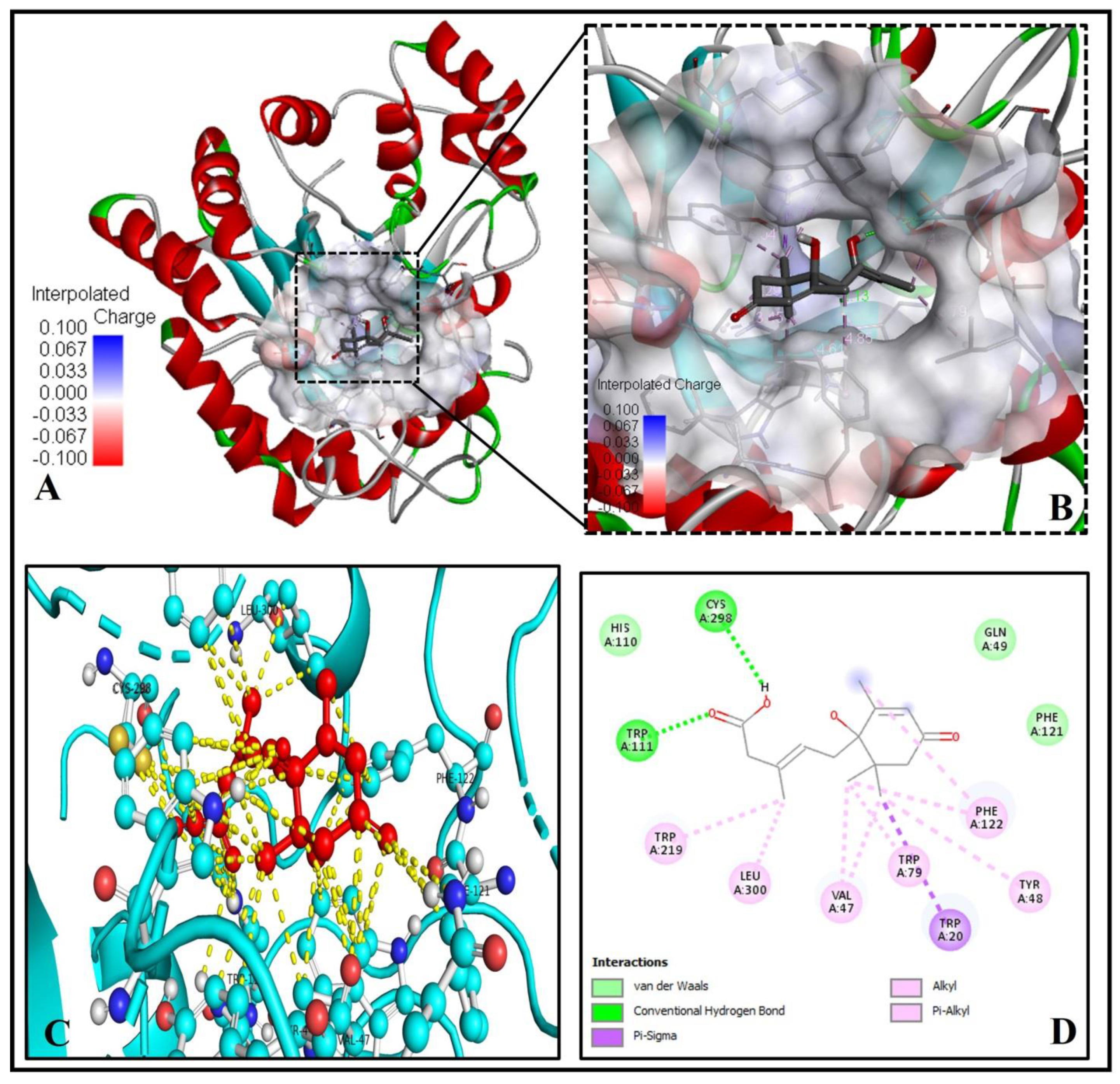
2.6. Analysis of the Molecular Binding Pattern of Abscisic Acid with Aldose Reductase
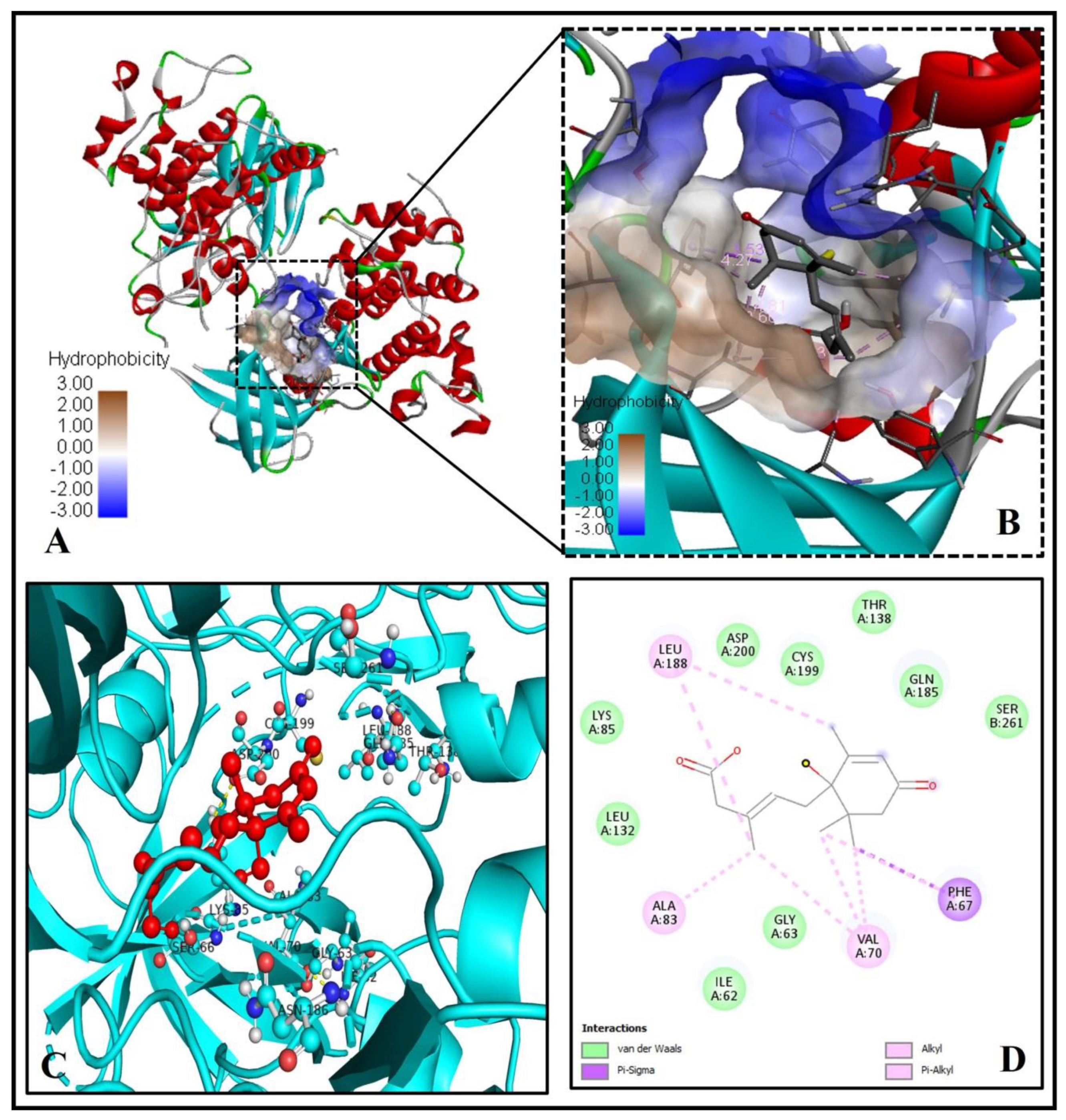
2.7. Analysis of Abscisic Acid and Glycogen Synthase Kinase-3 (GSK-3) Docked Complex
2.8. Screening of Pyruvate Dehydrogenase Kinase (PKD) and Abscisic Acid Docked Complex
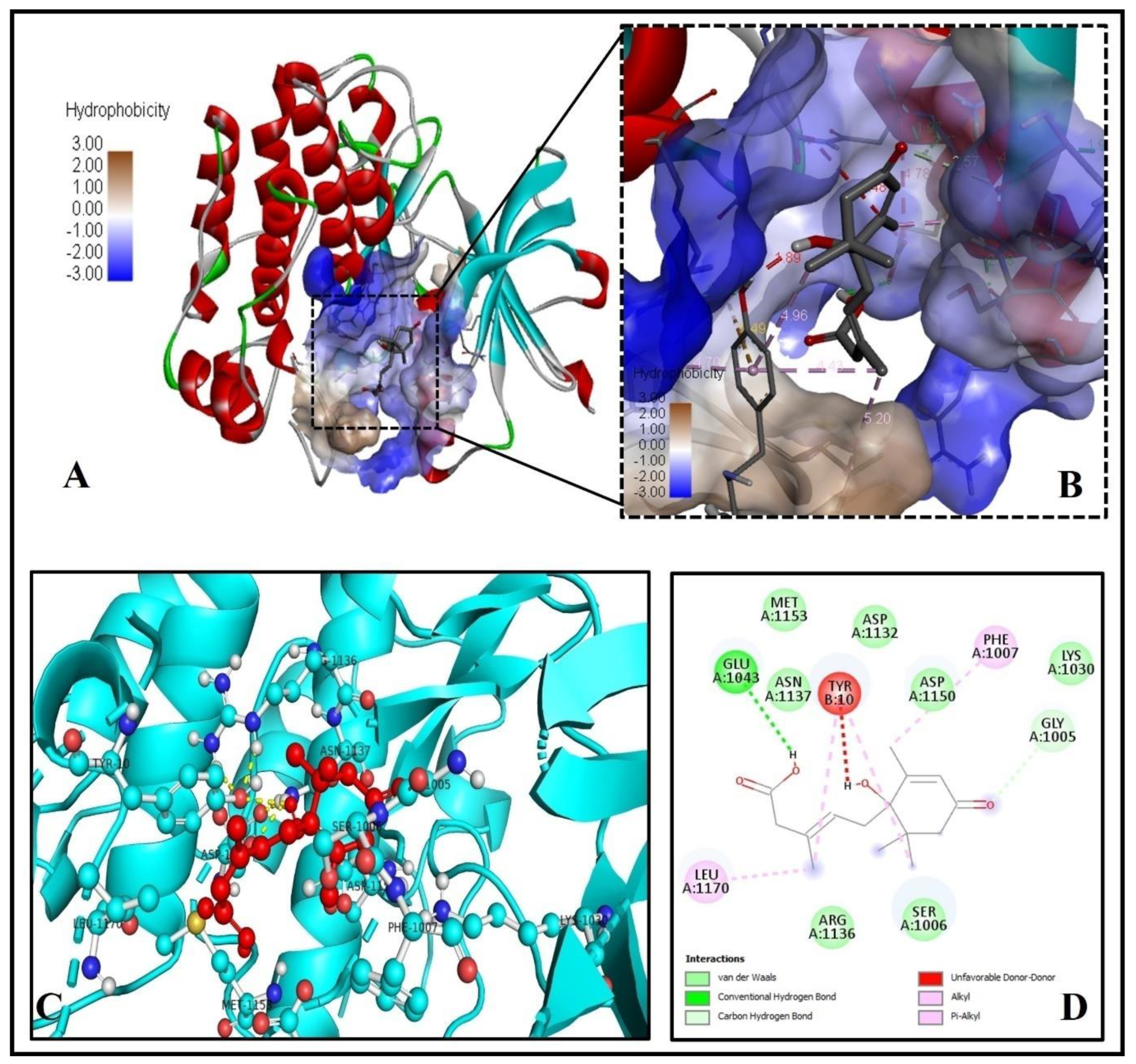
2.9. Investigation of the Docked Complex of Tyrosine Kinase with Abscisic Acid
2.10. Computational Pharmacodynamics Screening of Abscisic Acid Ligand
2.11. In Silico Pharmacokinetics and ADMET Evaluation of Abscisic Acid
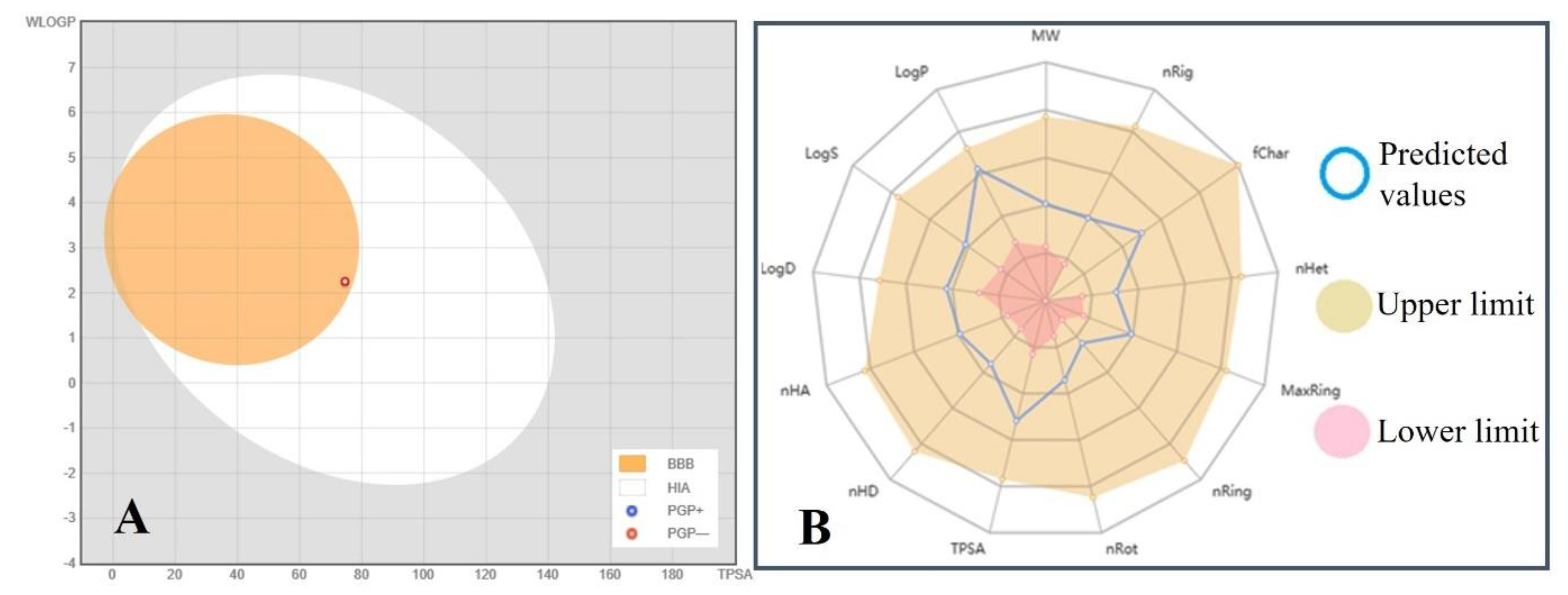
2.12. Boiled-Egg Plot and Radar Graph Analysis
3. Materials and Methods
3.1. Retrieval and Preparation of Proteins and Ligand
3.2. Molecular Docking
3.3. Post-Docking Protein-Ligand Interaction Analysis
3.4. Calculation of Inhibition Constant
3.5. Screening of Ligand Abscisic Acid for Pharmacodynamics Properties
3.6. Screening of Ligand Abscisic Acid for Pharmacokinetics and Drug-Likeness
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ashraf, S.A.; Elkhalifa, A.E.O.; Siddiqui, A.J.; Patel, M.; Awadelkareem, A.M.; Snoussi, M.; Ashraf, M.S.; Adnan, M.; Hadi, S. Cordycepin for Health and Wellbeing: A Potent Bioactive Metabolite of an Entomopathogenic Cordyceps Medicinal Fungus and Its Nutraceutical and Therapeutic Potential. Molecules 2020, 25, 2735. [Google Scholar] [CrossRef]
- Unnikrishnan, R.; Misra, A. Infections and diabetes: Risks and mitigation with reference to India. Diabetes Metab. Syndr. 2020, 14, 1889–1894. [Google Scholar] [CrossRef]
- Damián-Medina, K.; Salinas-Moreno, Y.; Milenkovic, D.; Figueroa-Yáñez, L.; Marino-Marmolejo, E.; Higuera-Ciapara, I.; Vallejo-Cardona, A.; Lugo-Cervantes, E. In silico analysis of antidiabetic potential of phenolic compounds from blue corn (Zea mays L.) and black bean (Phaseolus vulgaris L.). Heliyon 2020, 6, e03632. [Google Scholar] [CrossRef]
- Hameed, I.; Masoodi, S.R.; Mir, S.A.; Nabi, M.; Ghazanfar, K.; Ganai, B.A. Type 2 diabetes mellitus: From a metabolic disorder to an inflammatory condition. World J. Diabetes 2015, 6, 598–612. [Google Scholar] [CrossRef]
- Elkhalifa, A.E.O.; Al-Shammari, E.; Adnan, M.; Alcantara, J.C.; Mehmood, K.; Eltoum, N.E.; Awadelkareem, A.M.; Khan, M.A.; Ashraf, S.A. Development and Characterization of Novel Biopolymer Derived from Abelmoschus esculentus L. Extract and Its Antidiabetic Potential. Molecules 2021, 26, 3609. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-M.; Xu, R.; Wang, H.; Chen, J.-Y.; Tu, Z.-C. Structural Properties, Bioactivities, and Applications of Polysaccharides from Okra [Abelmoschus esculentus (L.) Moench]: A Review. J. Agric. Food Chem. 2020, 68, 14091–14103. [Google Scholar] [CrossRef] [PubMed]
- Elkhalifa, A.E.O.; Alshammari, E.; Adnan, M.; Alcantara, J.C.; Awadelkareem, A.M.; Eltoum, N.E.; Mehmood, K.; Panda, B.P.; Ashraf, S.A. Okra (Abelmoschus Esculentus) as a Potential Dietary Medicine with Nutraceutical Importance for Sustainable Health Applications. Molecules 2021, 26, 696. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, A.; Lucarini, M.; Novellino, E.; Souto, E.B.; Daliu, P.; Santini, A. Abelmoschus esculentus (L.): Bioactive Components’ Beneficial Properties-Focused on Antidiabetic Role-For Sustainable Health Applications. Molecules 2018, 24, 38. [Google Scholar] [CrossRef] [Green Version]
- Daliu, P.; Annunziata, G.; Tenore, G.C.; Santini, A. Abscisic acid identification in Okra, Abelmoschus esculentus L. (Moench): Perspective nutraceutical use for the treatment of diabetes. Nat. Prod. Res. 2020, 34, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Perez, A.M. Abscisic acid, a promising therapeutic molecule to prevent Alzheimer’s and neurodegenerative diseases. Neural Regen. Res. 2020, 15, 1035–1036. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Cackowski, F.C.; Yumoto, K.; Decker, A.M.; Wang, Y.; Hotchkin, M.; Lee, E.; Buttitta, L.; Taichman, R.S. Abscisic acid regulates dormancy of prostate cancer disseminated tumor cells in the bone marrow. Neoplasia 2021, 23, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.W.; Walker, E.A.; Bujalska, I.J.; Draper, N.; Lavery, G.G.; Cooper, M.S.; Hewison, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 1: A tissue-specific regulator of glucocorticoid response. Endocr. Rev. 2004, 25, 831–866. [Google Scholar] [CrossRef] [PubMed]
- Morton, N.M.; Paterson, J.M.; Masuzaki, H.; Holmes, M.C.; Staels, B.; Fievet, C.; Walker, B.R.; Flier, J.S.; Mullins, J.J.; Seckl, J.R. Novel adipose tissue-mediated resistance to diet-induced visceral obesity in 11 beta-hydroxysteroid dehydrogenase type 1-deficient mice. Diabetes 2004, 53, 931–938. [Google Scholar] [CrossRef] [Green Version]
- Morgan, S.A.; Sherlock, M.; Gathercole, L.L.; Lavery, G.G.; Lenaghan, C.; Bujalska, I.J.; Laber, D.; Yu, A.; Convey, G.; Mayers, R.; et al. 11beta-hydroxysteroid dehydrogenase type 1 regulates glucocorticoid-induced insulin resistance in skeletal muscle. Diabetes 2009, 58, 2506–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollis, G.; Huber, R. 11β-Hydroxysteroid dehydrogenase type 1 inhibition in type 2 diabetes mellitus. Diabetes Obes. Metab. 2011, 13, 1–6. [Google Scholar] [CrossRef]
- Alberts, P.; Nilsson, C.; Selen, G.; Engblom, L.O.; Edling, N.H.; Norling, S.; Klingström, G.; Larsson, C.; Forsgren, M.; Ashkzari, M.; et al. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology 2003, 144, 4755–4762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhme, T.; Engel, C.K.; Farjot, G.; Güssregen, S.; Haack, T.; Tschank, G.; Ritter, K. 1,1-Dioxo-5,6-dihydro-[1,2,4]oxathiazines, a novel class of 11ß-HSD1 inhibitors for the treatment of diabetes. Bioorganic Med. Chem. Lett. 2013, 23, 4685–4691. [Google Scholar] [CrossRef]
- Ryu, J.H.; Kim, S.; Lee, J.A.; Han, H.Y.; Son, H.J.; Lee, H.J.; Kim, Y.H.; Kim, J.S.; Park, H.G. Synthesis and optimization of picolinamide derivatives as a novel class of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Shiraki, R.; Sasuga, D.; Hosaka, M.; Kawano, T.; Fukudome, H.; Kurosawa, K.; Moritomo, A.; Mimasu, S.; Ishii, H.; et al. Discovery and Biological Evaluation of Potent and Orally Active Human 11β-Hydroxysteroid Dehydrogenase Type 1 Inhibitors for the Treatment of Type 2 Diabetes Mellitus. Chem. Pharm. Bull. 2019, 67, 824–838. [Google Scholar] [CrossRef] [Green Version]
- Nakaishi, Y.; Bando, M.; Shimizu, H.; Watanabe, K.; Goto, F.; Tsuge, H.; Kondo, K.; Komatsu, M. Structural analysis of human glutamine: Fructose-6-phosphate amidotransferase, a key regulator in type 2 diabetes. FEBS Lett. 2009, 583, 163–167. [Google Scholar] [CrossRef] [Green Version]
- Shanak, S.; Saad, B.; Zaid, H. Metabolic and Epigenetic Action Mechanisms of Antidiabetic Medicinal Plants. Evid.-Based Complementary Altern. Med. ECAM 2019, 2019, 3583067. [Google Scholar] [CrossRef] [PubMed]
- Belete, T.M. A Recent Achievement In the Discovery and Development of Novel Targets for the Treatment of Type-2 Diabetes Mellitus. J. Exp. Pharmacol. 2020, 12, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsley, J.E.; Rutter, J. Nutrient sensing and metabolic decisions. Comp. Biochem. Physiol. Part. B Biochem. Mol. Biol. 2004, 139, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jia, Y.; Cooper, J.J.; Hale, T.; Zhang, Z.; Elbein, S.C. Common variants in glutamine: Fructose-6-phosphate amidotransferase 2 (GFPT2) gene are associated with type 2 diabetes, diabetic nephropathy, and increased GFPT2 mRNA levels. J. Clin. Endocrinol. Metab. 2004, 89, 748–755. [Google Scholar] [CrossRef] [Green Version]
- Buse, M.G. Hexosamines, insulin resistance, and the complications of diabetes: Current status. Am. J. Physiol. Endocrinol. Metab. 2006, 290, E1–E8. [Google Scholar] [CrossRef]
- Savage, D.B.; Sewter, C.P.; Klenk, E.S.; Segal, D.G.; Vidal-Puig, A.; Considine, R.V.; O’Rahilly, S. Resistin/Fizz3 expression in relation to obesity and peroxisome proliferator-activated receptor-gamma action in humans. Diabetes 2001, 50, 2199–2202. [Google Scholar] [CrossRef] [Green Version]
- Salam, N.K.; Huang, T.H.; Kota, B.P.; Kim, M.S.; Li, Y.; Hibbs, D.E. Novel PPAR-gamma agonists identified from a natural product library: A virtual screening, induced-fit docking and biological assay study. Chem. Biol. Drug Des. 2008, 71, 57–70. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Saltiel, A.R. PPAR gamma and the treatment of insulin resistance. Trends Endocrinol. Metab. TEM 2000, 11, 362–368. [Google Scholar] [CrossRef]
- Staels, B. PPAR Agonists and the Metabolic Syndrome. Therapies 2007, 62, 319–326. [Google Scholar] [CrossRef]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef]
- Pan, P.W.; Feldman, J.L.; Devries, M.K.; Dong, A.; Edwards, A.M.; Denu, J.M. Structure and biochemical functions of SIRT6. J. Biol. Chem. 2011, 286, 14575–14587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kugel, S.; Mostoslavsky, R. Chromatin and beyond: The multitasking roles for SIRT6. Trends Biochem. Sci. 2014, 39, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gertman, O.; Omer, D.; Hendler, A.; Stein, D.; Onn, L.; Khukhin, Y.; Portillo, M.; Zarivach, R.; Cohen, H.Y.; Toiber, D.; et al. Directed evolution of SIRT6 for improved deacylation and glucose homeostasis maintenance. Sci. Rep. 2018, 8, 3538. [Google Scholar] [CrossRef] [PubMed]
- Blander, G.; Guarente, L. The Sir2 Family of Protein Deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agius, L. Targeting hepatic glucokinase in type 2 diabetes: Weighing the benefits and risks. Diabetes 2009, 58, 18–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Kabbani, O.; Ruiz, F.; Darmanin, C.; Chung, R.P. Aldose reductase structures: Implications for mechanism and inhibition. Cell. Mol. Life Sci. CMLS 2004, 61, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Kaul, C.L.; Ramarao, P. The role of aldose reductase inhibitors in diabetic complications: Recent trends. Methods Find. Exp. Clin. Pharmacol. 2001, 23, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Tso, S.C.; Qi, X.; Gui, W.J.; Wu, C.Y.; Chuang, J.L.; Wernstedt-Asterholm, I.; Morlock, L.K.; Owens, K.R.; Scherer, P.E.; Williams, N.S.; et al. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J. Biol. Chem. 2014, 289, 4432–4443. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2012, 64, 4–17. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Martin, Y.C. A bioavailability score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.rcsb.org/pdb/home (accessed on 15 April 2021).
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protein Data Bank in Europe. Available online: https://www.ebi.ac.uk/pdbe (accessed on 15 April 2021).
- PDBsum, Pictorial Database of 3D Structures in the Protein Data Bank. Available online: http://www.ebi.ac.uk/pdbsum (accessed on 17 April 2021).
- Pharmacokinetic Properties. Available online: http://biosig.unimelb.edu.au/pkcsm/prediction (accessed on 15 April 2021).
- SwissADME. Available online: http://www.swissadme.ch (accessed on 19 April 2021).
- ADMET Evaluation. Available online: https://admetmesh.scbdd.com/service/evaluation/cal (accessed on 19 April 2021).
- Zoete, V.; Daina, A.; Bovigny, C.; Michielin, O. SwissSimilarity: A Web Tool for Low to Ultra High Throughput Ligand-Based Virtual Screening. J. Chem. Inf. Model. 2016, 56, 1399–1404. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No | Protein Name (PDB ID) | Theoretical Weight (KDa) | Name of Chains | Binding Energy (ΔG) (kcal/mol) | Predicted Inhibition Constant pKi (µM) | No. of H-Bonds | H-Bond Forming Residues |
|---|---|---|---|---|---|---|---|
| 1 | 11β-HSD1 (4K1L) | 31.84 | A, B, C, D | −8.1 | 6.01 | 2 | TYR(A)183, SER(A)169 |
| 2. | GFAT (2ZJ4) | 42.32 | A | −7.3 | 5.21 | 5 | CYS373, THR375, GLN421, SER422, THR425 |
| 3. | PPAR-gamma (3DZY) | 51.53 | A, B, C, D | −7.3 | 5.21 | 2 | TYR(A)189, TYR(D)250 |
| 4. | SIRT6 (3K35) | 35.15 | A, B, C, D, E, F | −7.3 | 5.21 | 2 | GLN(C)111, HIS(C)131 |
| 5. | Glucokinase (4IXC) | 50.81 | A | −6.8 | 5.05 | 3 | ASN83, ARG85, GLY229 |
| 6. | Aldose reductase (3G5E) | 36.18 | A | −6.6 | 4.84 | 2 | TRP111, CYS298 |
| 7. | Glycogen synthase kinase-3 (3F7Z) | 39.88 | A, B | −6.6 | 4.84 | 0 | - |
| 8. | Pyruvate dehydrogenase kinase (4MP2) | 45.23 | A | −6.3 | 4.55 | 1 | AGR162 |
| 9. | Tyrosine kinase (1IR3) | 35.03 | A | −6.2 | 4.47 | 1 | GLU1043 |
| S. No | Parameters | Bioactivity Score |
|---|---|---|
| 1 | GPCR ligand | −0.01 ↓↓↓ |
| 2 | Ion channel modulator | 0.28 ↑ |
| 3 | Kinase inhibitor | −0.61 ↓ |
| 4 | Nuclear receptor ligand | 1.06 ↑↑↑ |
| 5 | Protease inhibitor | −0.20 ↓↓ |
| 6 | Enzyme inhibitor | 0.75 ↑↑ |
| Physicochemical Properties | Predicted Values | Absorption | Predicted Value | Distribution | Predicted Values | Metabolism | Predicted Value | Extraction and Toxicity | Predicted Value |
|---|---|---|---|---|---|---|---|---|---|
| LogP, LogS and LogD | 2.342, −2.465 and 1.656 | Water solubility logP | −2.253 mol/L | Volume distribution (VD) of a drug in blood plasmas | 0.343 L/kg | CYP2D6 substrate and CYP3A4 substrate | No | Total drug clearance log (CLtot) | 0.685 mL/min/kg |
| Molecular weight | 264.32 g/mol | Lipd solibility LogP | 1.96 Log Po/w | Plasma protein binding (PPB) | 79.896% | CYP2D6 inhibitor | No | Renal organic cation transporter (OCT2) substrate | No |
| Number of hydrogen bond acceptors (nHA), donors (nHD), and rotatable bonds (nRot) | 4, 2 and 3 | Caco2 permeability | 0.913 og Papp in 10–6 cm/s | The fraction unbound in blood plasmas (Fu) | 9.191% | CYP3A4 inhibitor | No | AMES toxicity, hepatotoxicity, skin sensitization, hERG I & II inhibitor | No |
| Number of rings (nRing), rigid bonds (nRig), heteroatoms (nHet), and atoms in the biggest ring (MaxRing) | 1, 10, 4 and 6 | Log Kp skin permeability | −2.715 cm/s | BBB permeability | −0.047 log BB | CYP1A2 inhibitor | No | Max. tolerated dose (human) | 0.304 log mg/kg/day |
| Formal charge (fChar) | 0 | Human intestinal absorption (HIA) | 96.712% | CNS permeability | −2.913 log PS | CYP2C19 inhibitor | No | Oral rat acute toxicity (LD50) | 1.793 mol/kg |
| Molecular total polar surface area (TPSA) | 74.60 Ų | P-glycoprotein substrate, P-glycoprotein I & II inhibitor | No | Bioavailability score | 0.85 | CYP2C9 inhibitor | No | T. Pyriformis toxicity | 0.42 log ug/L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashraf, S.A.; Elkhalifa, A.E.O.; Mehmood, K.; Adnan, M.; Khan, M.A.; Eltoum, N.E.; Krishnan, A.; Baig, M.S. Multi-Targeted Molecular Docking, Pharmacokinetics, and Drug-Likeness Evaluation of Okra-Derived Ligand Abscisic Acid Targeting Signaling Proteins Involved in the Development of Diabetes. Molecules 2021, 26, 5957. https://doi.org/10.3390/molecules26195957
Ashraf SA, Elkhalifa AEO, Mehmood K, Adnan M, Khan MA, Eltoum NE, Krishnan A, Baig MS. Multi-Targeted Molecular Docking, Pharmacokinetics, and Drug-Likeness Evaluation of Okra-Derived Ligand Abscisic Acid Targeting Signaling Proteins Involved in the Development of Diabetes. Molecules. 2021; 26(19):5957. https://doi.org/10.3390/molecules26195957
Chicago/Turabian StyleAshraf, Syed Amir, Abd Elmoneim O. Elkhalifa, Khalid Mehmood, Mohd Adnan, Mushtaq Ahmad Khan, Nagat Elzein Eltoum, Anuja Krishnan, and Mirza Sarwar Baig. 2021. "Multi-Targeted Molecular Docking, Pharmacokinetics, and Drug-Likeness Evaluation of Okra-Derived Ligand Abscisic Acid Targeting Signaling Proteins Involved in the Development of Diabetes" Molecules 26, no. 19: 5957. https://doi.org/10.3390/molecules26195957
APA StyleAshraf, S. A., Elkhalifa, A. E. O., Mehmood, K., Adnan, M., Khan, M. A., Eltoum, N. E., Krishnan, A., & Baig, M. S. (2021). Multi-Targeted Molecular Docking, Pharmacokinetics, and Drug-Likeness Evaluation of Okra-Derived Ligand Abscisic Acid Targeting Signaling Proteins Involved in the Development of Diabetes. Molecules, 26(19), 5957. https://doi.org/10.3390/molecules26195957