
Metal–Metal Bond in the Light of Pauling’s Rules



Abstract
:1. Introduction
2. Methods: Determination of the BV Parameters for the Metal–Metal Bonds
3. Results and Discussion
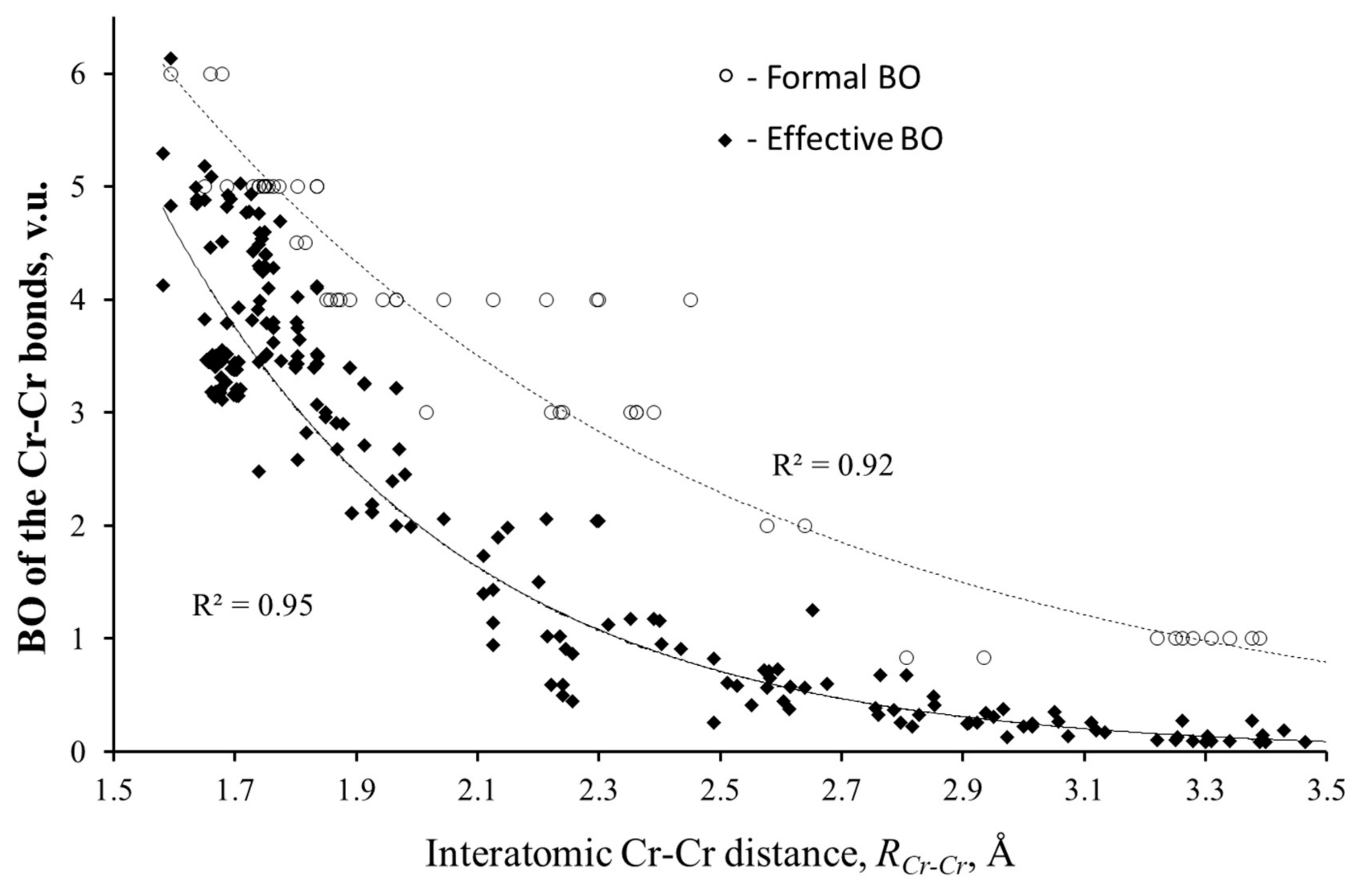
3.1. Validity of Equation (2) for Localized and Delocalized Bonds
3.2. Validity of Equation (1) for Compounds with Metal–Metal Bonds
3.2.1. Stretching of Metal–Metal Bonds Evident by Valence Violations
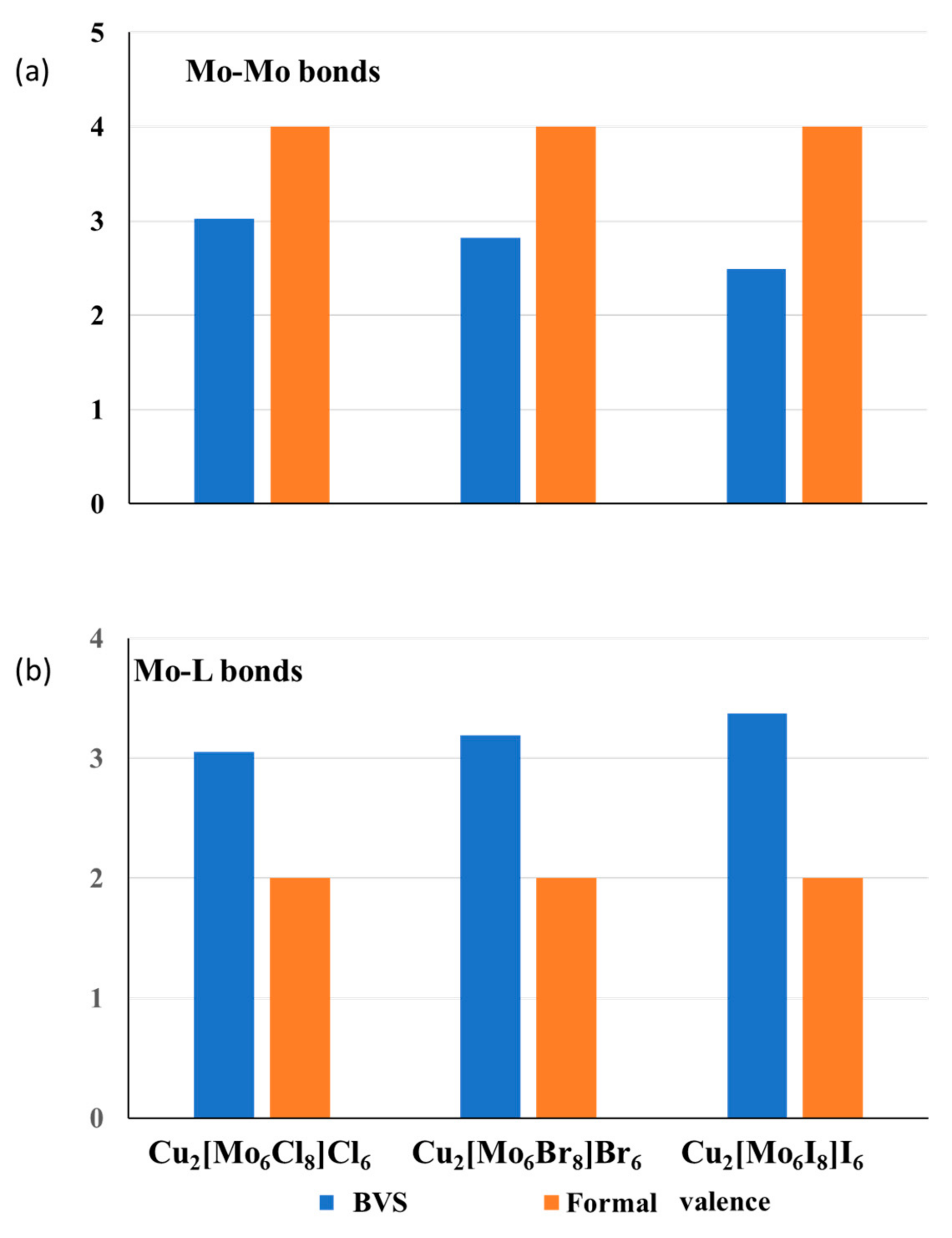
3.2.2. Compensation of the Valence Violations for the Bonds around Transition Metals
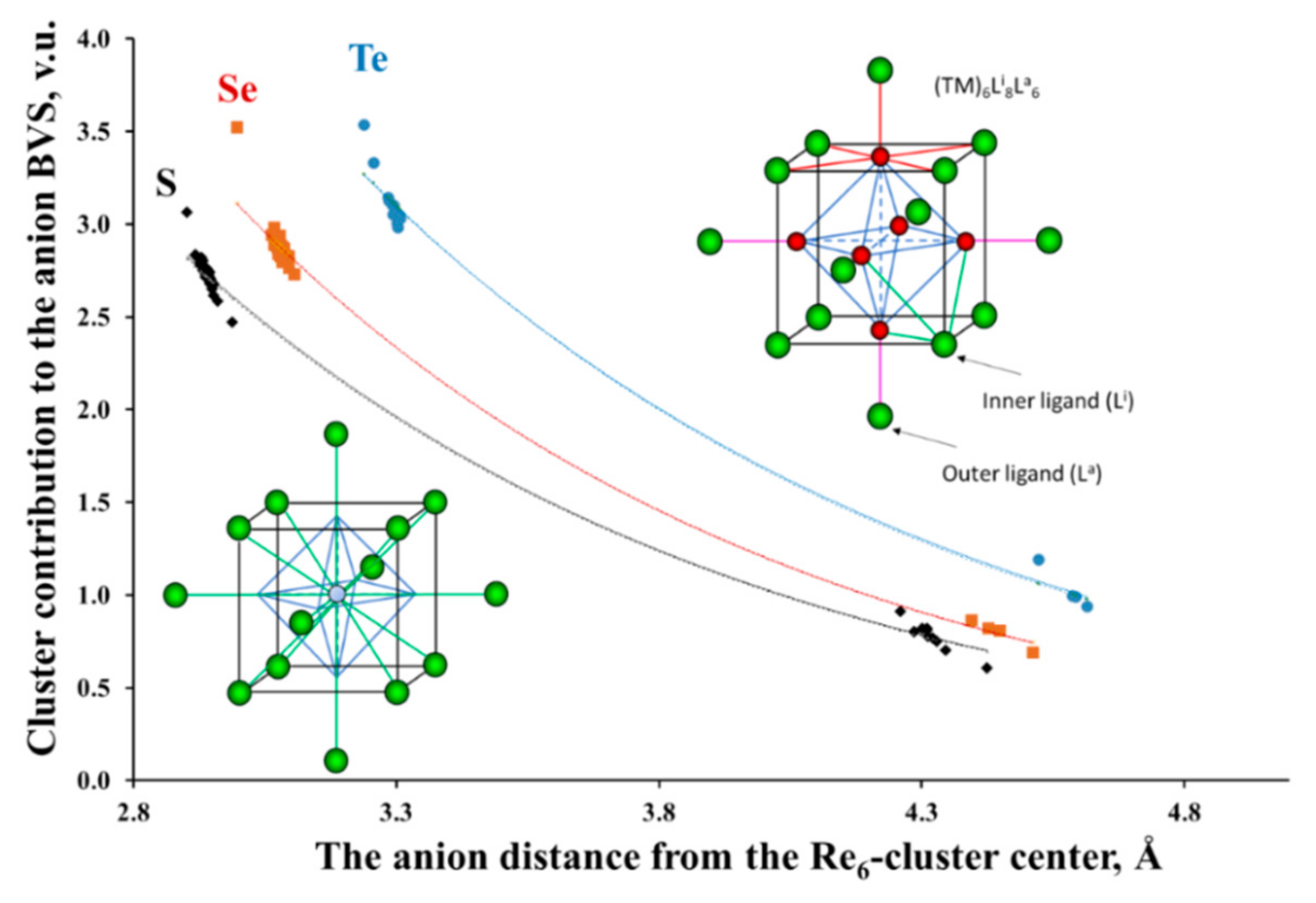
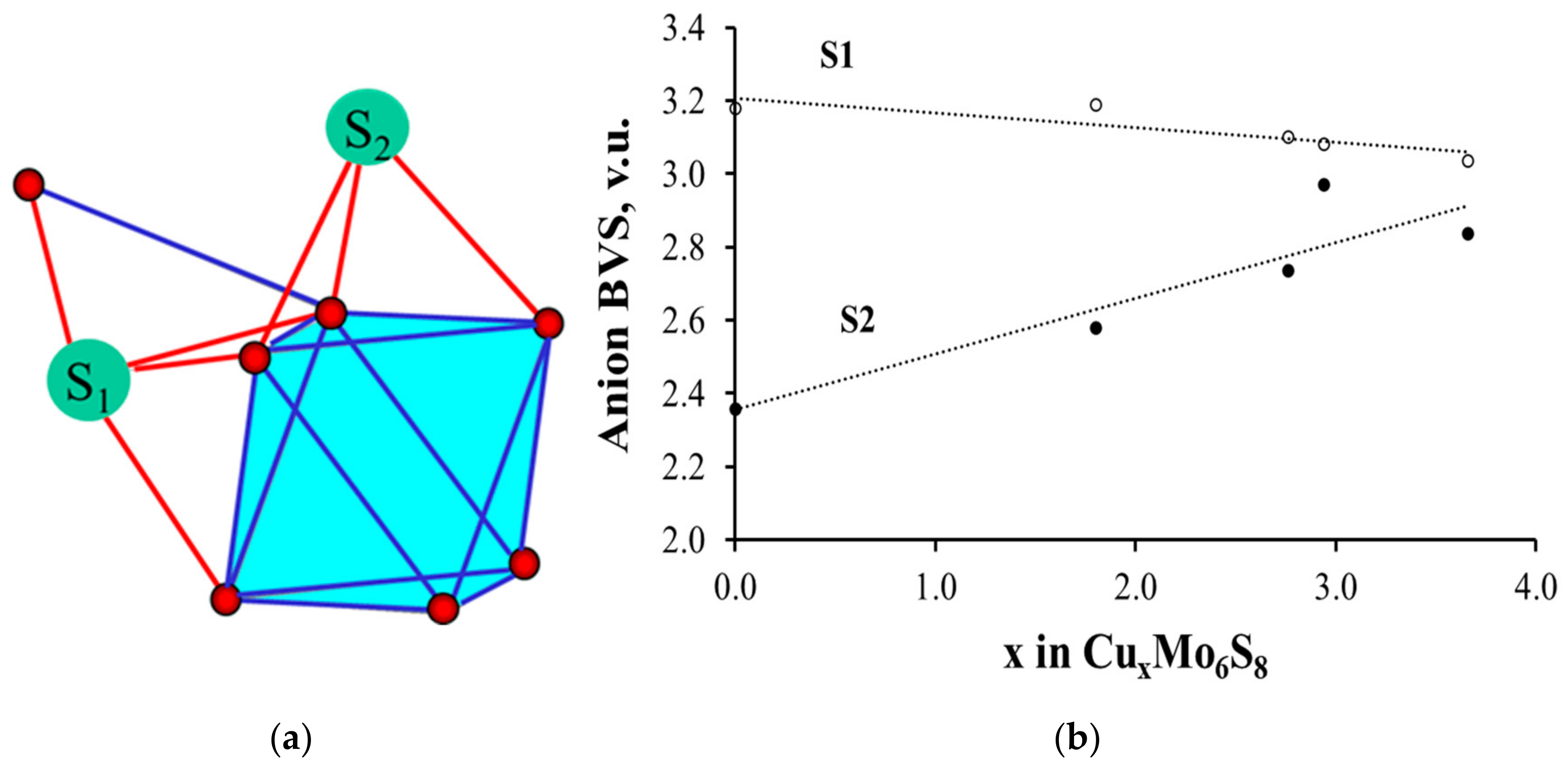
3.2.3. Clusters as Single Cations with Nonuniform BVS Distribution on Their Ligands
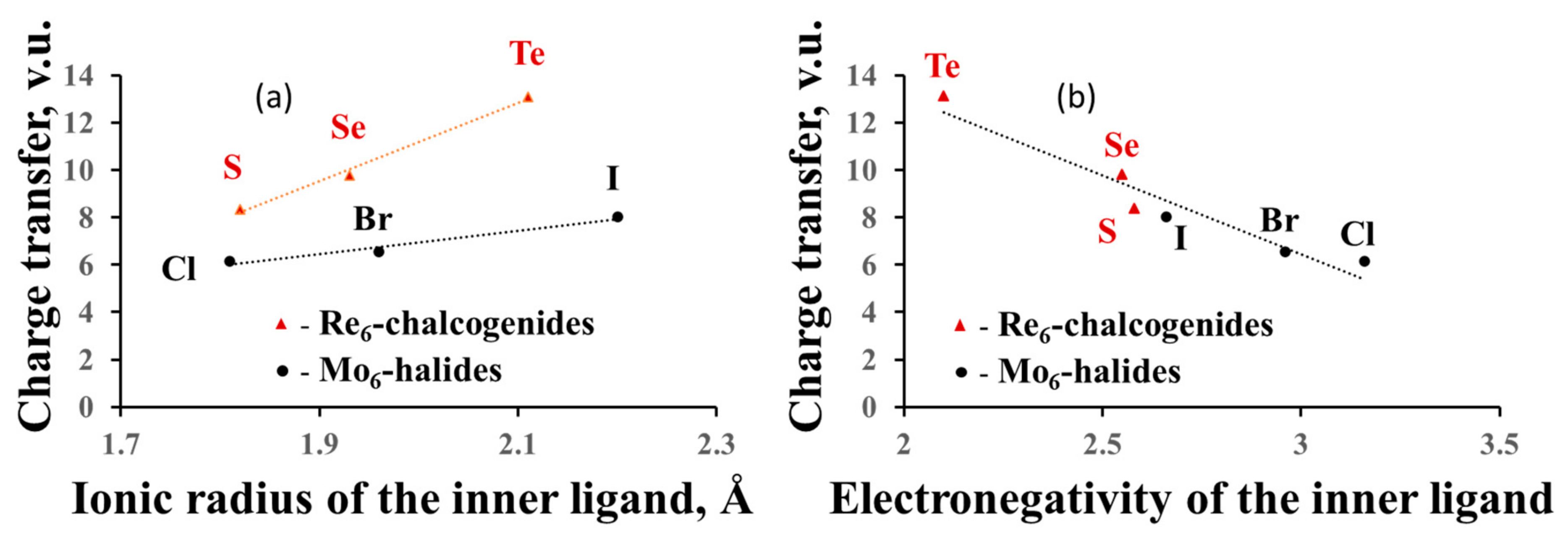
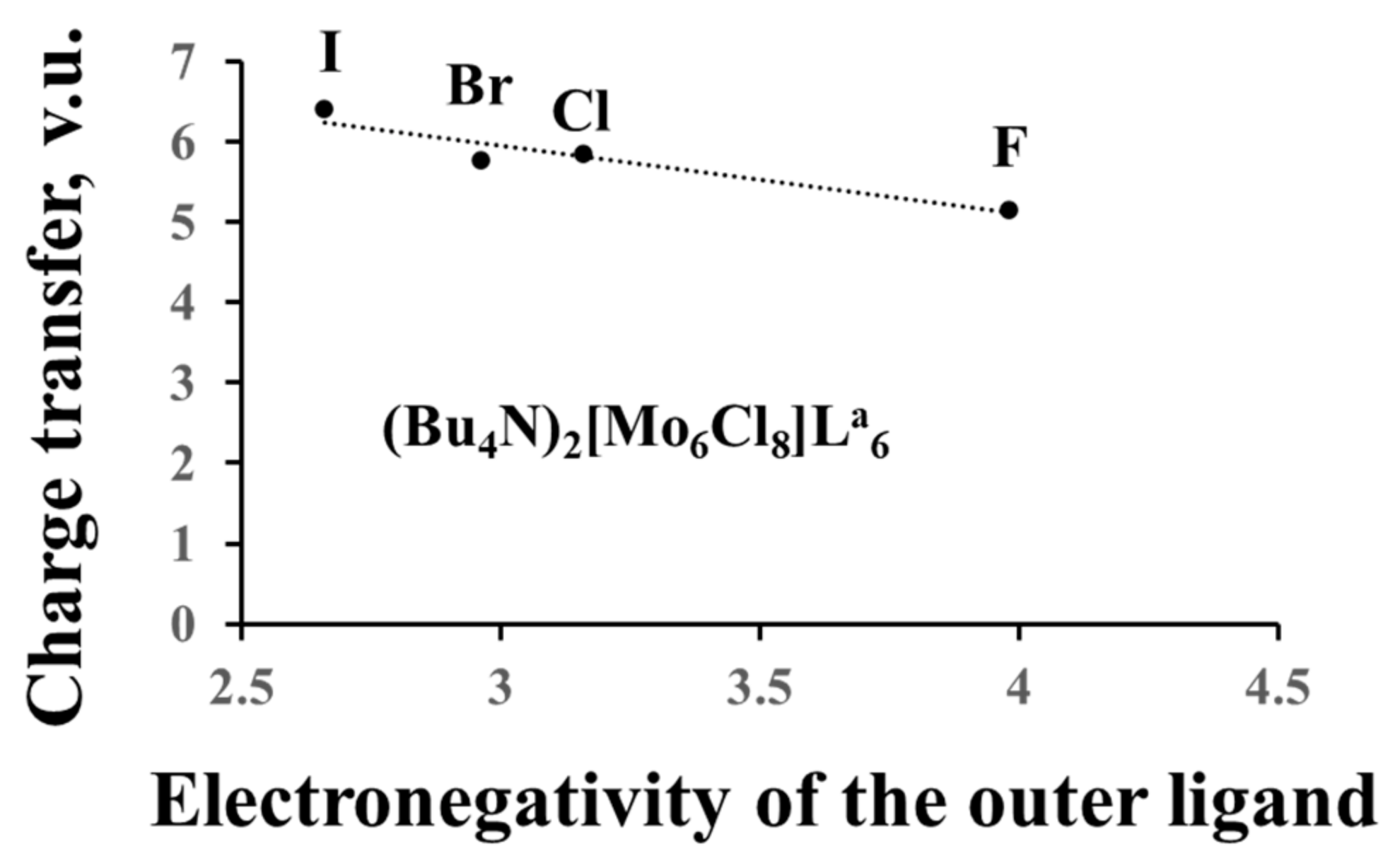
3.2.4. Charge Transfer from the Cluster to the Ligands and between the Ligands
3.2.5. Ligand Valence Violations as a Source of Material Instability
3.3. Comparison with the Results of Quantum Chemistry Calculations
3.4. The BVM Application to the Electrode Materials with Metal–Metal Bonding
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pauling, L. The principles determining the structure of complex ionic crystals. J. Am. Chem. Soc. 1929, 51, 1010–1026. [Google Scholar] [CrossRef]
- Pauling, L. Atomic Radii and Interatomic Distances in Metals. J. Am. Chem. Soc. 1947, 69, 542–553. [Google Scholar] [CrossRef]
- Brown, I.D. The Chemical Bond. In Inorganic Chemistry: The Bond. Valence Model; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Brown, I.D. Recent developments in the methods and applications of the bond valence model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, I.D. Available online: https://www.iucr.org/resources/data/datasets/bond-valence-parameters (accessed on 3 November 2016).
- George, J.; Waroquiers, D.; Di Stefano, D.; Petretto, G.; Rignanese, G.-M.; Hautier, G. The Limited Predictive Power of the Pauling Rules. Angew. Chem. Int. Ed. 2020, 59, 7569–7575. [Google Scholar] [CrossRef] [Green Version]
- Cotton, F.A.; Murillo, C.A.; Walton, R.A. Multiple Bonds between Metal Atoms; Springer: New York, NY, USA, 2005. [Google Scholar]
- Levi, E.; Aurbach, D. Lattice strains in the ligand framework in the octahedral metal cluster compounds as the origin of their instability. Chem. Mater. 2011, 23, 1901–1914. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Isnard, O. Bond-valence model for metal cluster compounds. I. Common lattice strains. Acta Cryst. B 2013, 69, 419–425. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Isnard, O. Bond-valence model for metal cluster compounds. II. Matrix effect. Acta Cryst. B 2013, 69, 426–438. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Isnard, O. Electronic effect related to the non-uniform distribution of ionic charges in metal cluster chalcogenide-halides. Eur. J. Inorg. Chem. 2014, 2014, 3736–3746. [Google Scholar] [CrossRef]
- Singh, V.; Dixit, M.; Kosa, M.; Major, D.T.; Levi, E.; Aurbach, D. Is it True that the Normal Valence-Length Correlation is Irrelevant for Metal-Metal Bonds? Chem. Eur. J. 2016, 22, 5269–5276. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Gatti, C. Do the Basic Crystal Chemistry Principles Agree with a Plethora of Recent Quantum Chemistry Data? IUCrJ 2018, 5, 542–547. [Google Scholar] [CrossRef] [Green Version]
- Levi, E.; Aurbach, D.; Gatti, C. Bond Order Conservation Principle and Peculiarities of the Metal-Metal Bonding. Inorg. Chem. 2018, 57, 15550–15557. [Google Scholar] [CrossRef] [PubMed]
- Levi, E.; Aurbach, D.; Gatti, C. Assessing the Strength of Metal–Metal Interactions. Inorg. Chem. 2019, 58, 7466–7471. [Google Scholar] [CrossRef] [PubMed]
- Levi, E.; Aurbach, D.; Gatti, C. Steric and Electrostatic Effects in Compounds with Centered Clusters Quantified by Bond Order Analysis. Cryst. Growth Des. 2020, 20, 2115–2122. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D.; Gatti, C. A revisit of the bond valence model makes it universal. PCCP 2020, 22, 13839–13849. [Google Scholar] [CrossRef] [PubMed]
- Burdett, J.K.; McLarnan, T.J. An orbital interpretation of Pauling’s rules. Am. Mineral. 1984, 69, 601–621. [Google Scholar]
- Mohri, F. Molecular orbital study of the bond-valence sum rule using Lewis-electron pair theory. Acta Cryst. B 2003, 59, 190–208. [Google Scholar] [CrossRef]
- Mohri, F. Molecular orbital study of bond–valence sum rule for hydrogen bond systems using Lewis-electron pair theory. J. Mol. Struct. Theochem. 2005, 756, 25–33. [Google Scholar] [CrossRef]
- Hardcastle, F.; Laffoon, S. Theoretical Justification for Bond Valence--Bond Length Empirical Correlations. JAAS 2012, 66, 87–93. [Google Scholar]
- Adams, S. Practical considerations in determining bond valence parameters. In Bond Valences; Springer: Berlin, Germany, 2013; pp. 91–128. [Google Scholar]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Cryst. B 1991, 47, 192–197. [Google Scholar] [CrossRef]
- O’Keefe, M.; Brese, N. Atom sizes and bond lengths in molecules and crystals. J. Am. Chem. Soc. 1991, 113, 3226–3229. [Google Scholar] [CrossRef]
- Wu, L.-C.; Hsu, C.-W.; Chuang, Y.-C.; Lee, G.-H.; Tsai, Y.-C.; Wang, Y. Bond characterization on a Cr–Cr quintuple bond: A combined experimental and theoretical study. J. Phys. Chem. A 2011, 115, 12602–12615. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Borin, A.C.; Gagliardi, L. Reaching the maximum multiplicity of the covalent chemical bond. Angew. Chem. 2007, 119, 1491–1494. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.D. Chemical and steric constraints in inorganic solids. Acta Cryst. B 1992, 48, 553–572. [Google Scholar] [CrossRef]
- Levi, E.; Aurbach, D. Lattice strains in the layered Mn, Ni and Co oxides as cathode materials in Li and Na batteries. Solid State Ion. 2014, 264, 54–68. [Google Scholar] [CrossRef]
- Schäfer, H.; Schnering, H.V. Metall-metall-bindungen bei niederen halogeniden, oxyden und oxydhalogeniden schwerer übergangsmetalle thermochemische und strukturelle prinzipien. Angew. Chem. 1964, 76, 833–849. [Google Scholar] [CrossRef]
- Corbett, J.D. Correlation of metal-metal bonding in halides and chalcides of the early transition elements with that in the metals. J. Solid State Chem. 1981, 37, 335–351. [Google Scholar] [CrossRef]
- Corbett, J.D. Chevrel phases: An analysis of their metal-metal bonding and crystal chemistry. J. Solid State Chem. 1981, 39, 56–74. [Google Scholar] [CrossRef]
- Prokopuk, N.; Shriver, D. The Octahedral M6Y8 and M6Y12 Clusters of Group 4 and 5 Transition Metals. In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press: San Diego, CA, USA, 1998; Volume 46, pp. 1–49. [Google Scholar]
- Adams, S. Relationship between bond valence and bond softness of alkali halides and chalcogenides. Acta Cryst. B 2001, 57, 278–287. [Google Scholar] [CrossRef]
- Baranovski, V.I.; Korolkov, D.V. Electron density distribution, polarizability and bonding in the octahedral clusters [Mo6S8(CN)6]6−, [Re6S8(CN)6]4− and Rh6(CO)16. Polyhedron 2004, 23, 1519–1526. [Google Scholar] [CrossRef]
- Yvon, K. Bonding and relationships between structure and physical properties in Chevrel-phase compounds MxMo6X8 (M = metal, x = S, Se, Te). In Current Topics in Materials Science; Kaldis, E., Ed.; Elsevier: Amsterdam, The Netherlands, 1979; Volume 3, pp. 53–129. [Google Scholar]
- Fischer, O.; Maple, M.B. (Eds.) Superconductivity in Ternary Compounds I; Springer: Berlin, Germany, 1982; Volume 32. [Google Scholar]
- Caillat, T.; Fleurial, J.P.; Snyder, G.J. Potential of Chevrel phases for thermoelectric applications. Solid State Sci. 1999, 1, 535–544. [Google Scholar] [CrossRef]
- Peña, O. Chevrel phases: Past, present and future. Physica C 2015, 514, 95–112. [Google Scholar] [CrossRef]
- Aurbach, D.; Lu, Z.; Schechter, A.; Gofer, Y.; Gizbar, H.; Turgeman, R.; Cohen, Y.; Moshkovich, M.; Levi, E. Prototype systems for rechargeable magnesium batteries. Nature 2000, 407, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Levi, E.; Gofer, Y.; Aurbach, D. On the way to rechargeable Mg batteries: The challenge of new cathode materials. Chem. Mater. 2009, 22, 860–868. [Google Scholar] [CrossRef]
- Levi, E.; Gershinsky, G.; Aurbach, D.; Isnard, O.; Ceder, G. New insight on the unusually high ionic mobility in chevrel phases. Chem. Mater. 2009, 21, 1390–1399. [Google Scholar] [CrossRef]
- Levi, E.; Lancry, E.; Mitelman, A.; Aurbach, D.; Ceder, G.; Morgan, D.; Isnard, O. Phase Diagram of Mg Insertion into Chevrel Phases, MgxMo6T8 (T = S, Se). 1. Crystal Structure of the Sulfides. Chem. Mater. 2006, 18, 5492–5503. [Google Scholar] [CrossRef]
- Thöle, F.; Wan, L.F.; Prendergast, D. Re-examining the Chevrel phase Mo6S8 cathode for Mg intercalation from an electronic structure perspective. PCCP 2015, 17, 22548–22551. [Google Scholar] [CrossRef]
- Wan, L.F.; Wright, J.; Perdue, B.R.; Fister, T.T.; Kim, S.; Apblett, C.A.; Prendergast, D. Revealing electronic structure changes in Chevrel phase cathodes upon Mg insertion using X-ray absorption spectroscopy. PCCP 2016, 18, 17326–17329. [Google Scholar] [CrossRef]
- Yu, P.; Long, X.; Zhang, N.; Feng, X.; Fu, J.; Zheng, S.; Ren, G.; Liu, Z.; Wang, C.; Liu, X. Charge Distribution on S and Intercluster Bond Evolution in Mo6S8 during the Electrochemical Insertion of Small Cations Studied by X-ray Absorption Spectroscopy. J. Phys. Chem. Lett. 2019, 10, 1159–1166. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Corbett, 1981 | Levi et al, 2019 | ||
|---|---|---|---|---|
| R0, Å | b, Å | R0, Å | b, Å | |
| Sc-Sc | 2.921 | 0.26 | 2.695 | 0.28 |
| Ti-Ti | 2.638 | 0.26 | 2.505 | 0.47 |
| V-V | 2.464 | 0.26 | 2.435 | 0.43 |
| Fe-Fe | 2.367 | 0.26 | 2.26 | 0.44 |
| Co-Co | 2.323 | 0.26 | 2.11 | 0.44 |
| Zr-Zr | 2.918 | 0.26 | 2.89 | 0.43 |
| Nb-Nb | 2.708 | 0.26 | 2.64 | 0.395 |
| Mo-Mo | 2.619 | 0.26 | 2.51 | 0.34 |
| W-W | 2.635 | 0.26 | 2.535 | 0.29 |
| Bond | R0TM6-L, Å | bTM6-L, Å | Bond | R0TM6-L, Å | bTM6-L, Å |
|---|---|---|---|---|---|
| Nb6-F | 2.96 | 1.36 | |||
| Nb6-Cl | 3.66 | 1.07 | W6-Cl | 3.79 | 1.38 |
| Nb6-Br | 3.925 | 1.01 | W6-Br | 3.96 | 1.18 |
| Nb6-I | 4.2 | 1.12 | W6-I | 4.22 | 1.08 |
| Mo6-Cl | 3.75 | 1.25 | Re6-S | 4.03 | 1.09 |
| Mo6-Br | 3.905 | 1.14 | Re6-Se | 4.2 | 1.06 |
| Mo6-I | 4.17 | 1.07 | Re6-Te | 4.59 | 1.14 |
| Atom | Bonds | R, Å | BO, vu | BVS, vu | Formal Valence, vu | Valence Violations, vu |
|---|---|---|---|---|---|---|
| Mo | Mo-Mo | 2.735 | 0.637 | 2.548 | 3.333 | 0.785 |
| Mo-L | 3.381 | 2.667 | −0.714 | |||
| All | 5.929 | 6 | 0.071 | |||
| S | Mo-S | 2.539 | 0.731 | 2.193 | −2 | −0.193 |
| C | Mo-C | 2.305 | 0.457 | 0.457 | −1 | 0.543 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levi, E.; Aurbach, D.; Gatti, C. Metal–Metal Bond in the Light of Pauling’s Rules. Molecules 2021, 26, 304. https://doi.org/10.3390/molecules26020304
Levi E, Aurbach D, Gatti C. Metal–Metal Bond in the Light of Pauling’s Rules. Molecules. 2021; 26(2):304. https://doi.org/10.3390/molecules26020304
Chicago/Turabian StyleLevi, Elena, Doron Aurbach, and Carlo Gatti. 2021. "Metal–Metal Bond in the Light of Pauling’s Rules" Molecules 26, no. 2: 304. https://doi.org/10.3390/molecules26020304
APA StyleLevi, E., Aurbach, D., & Gatti, C. (2021). Metal–Metal Bond in the Light of Pauling’s Rules. Molecules, 26(2), 304. https://doi.org/10.3390/molecules26020304