
Synthesis and Biochemical Evaluation of Warhead-Decorated Psoralens as (Immuno)Proteasome Inhibitors

 , , ,
, , ,
Abstract
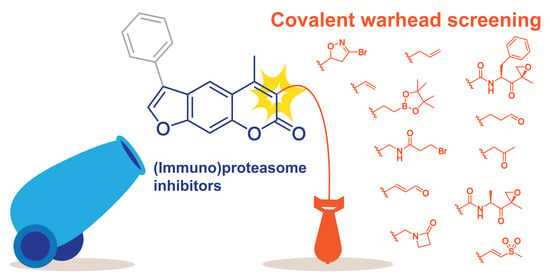
:
1. Introduction
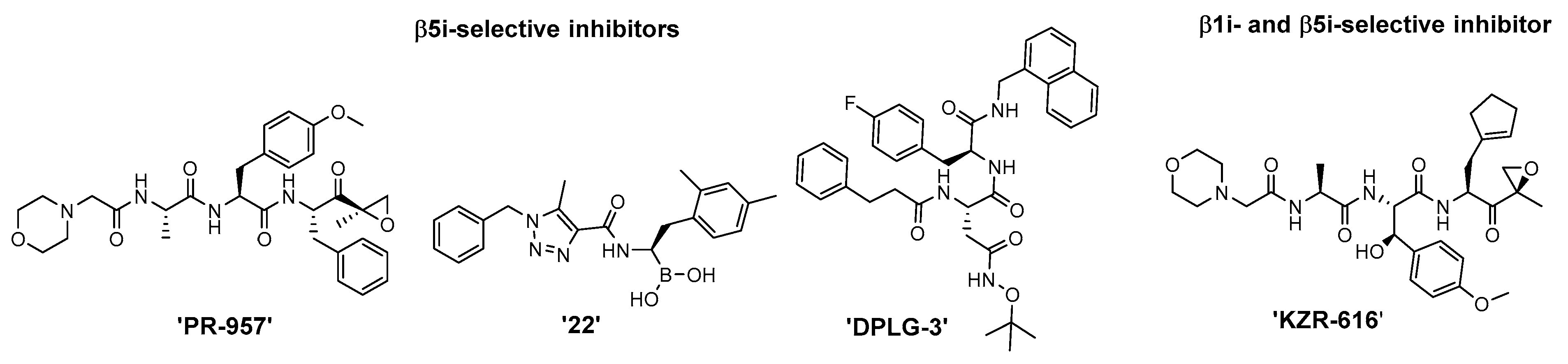
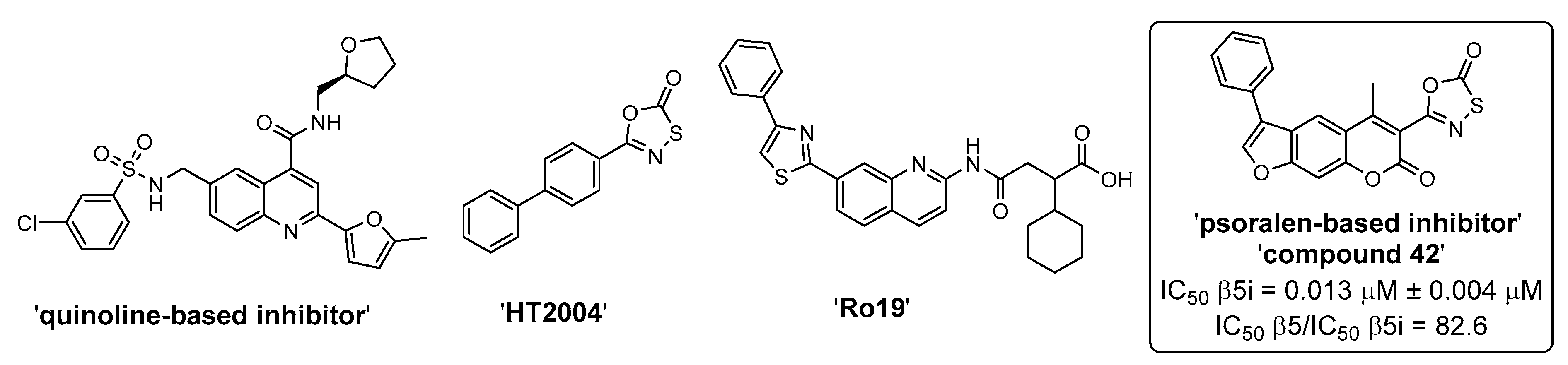
2. Results and Discussion
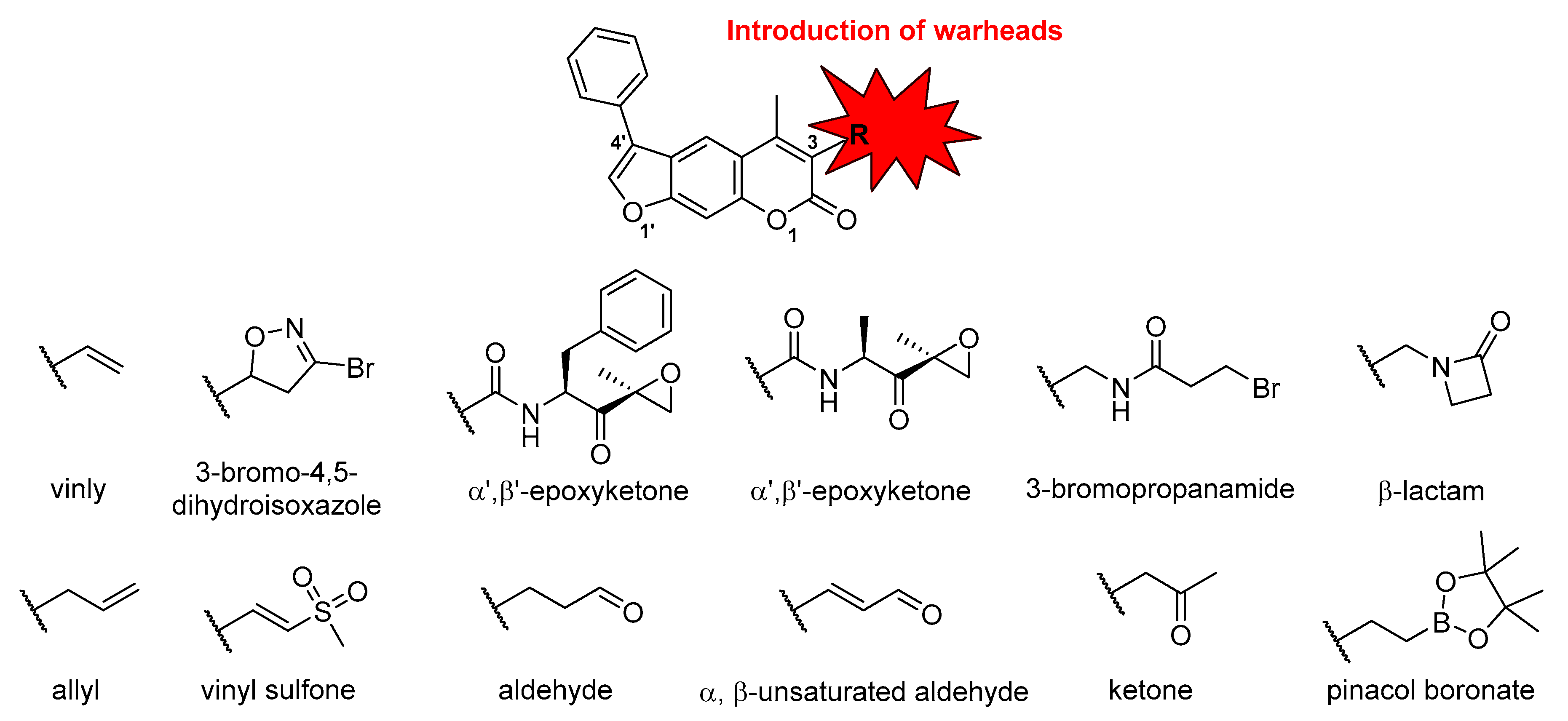
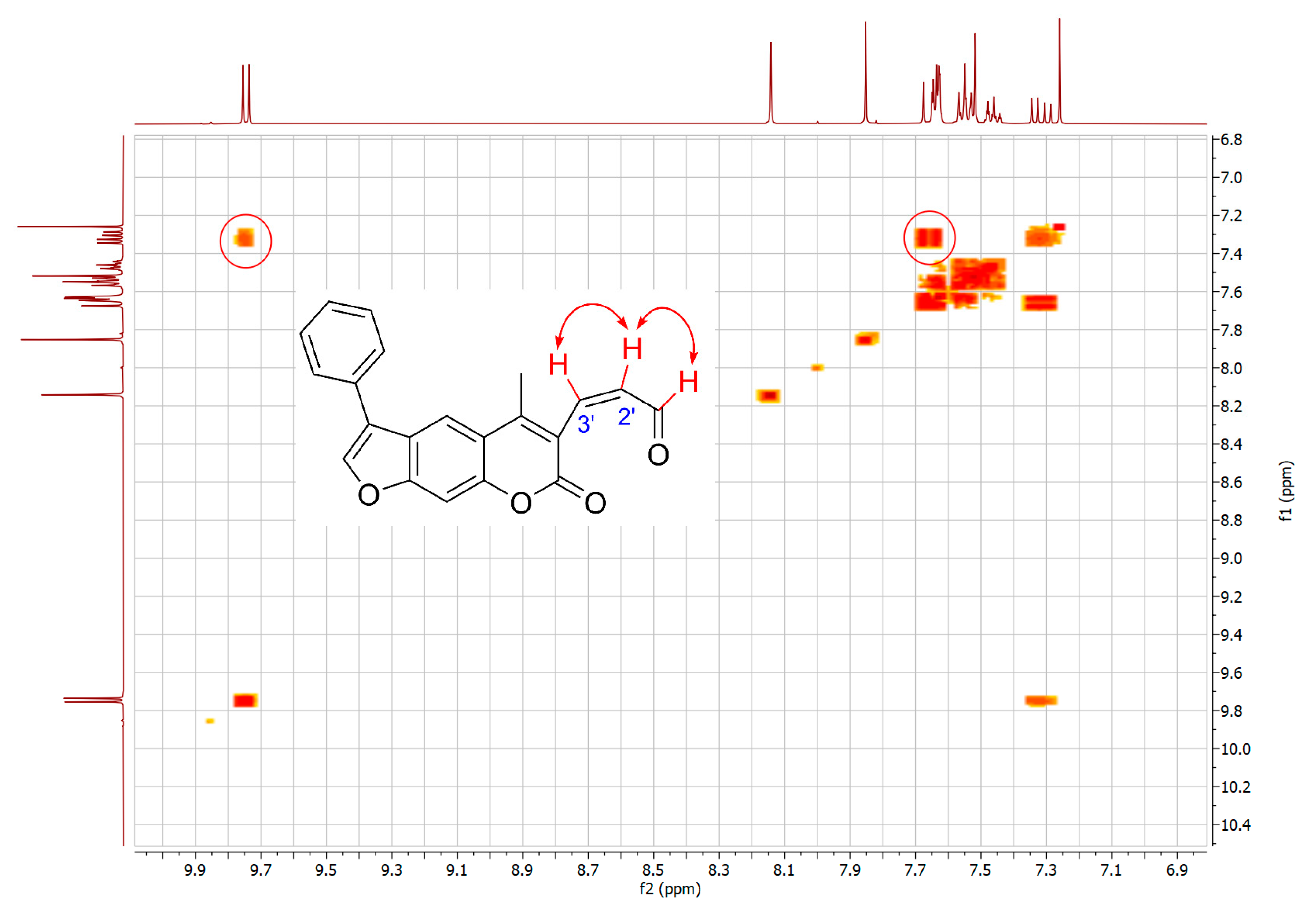
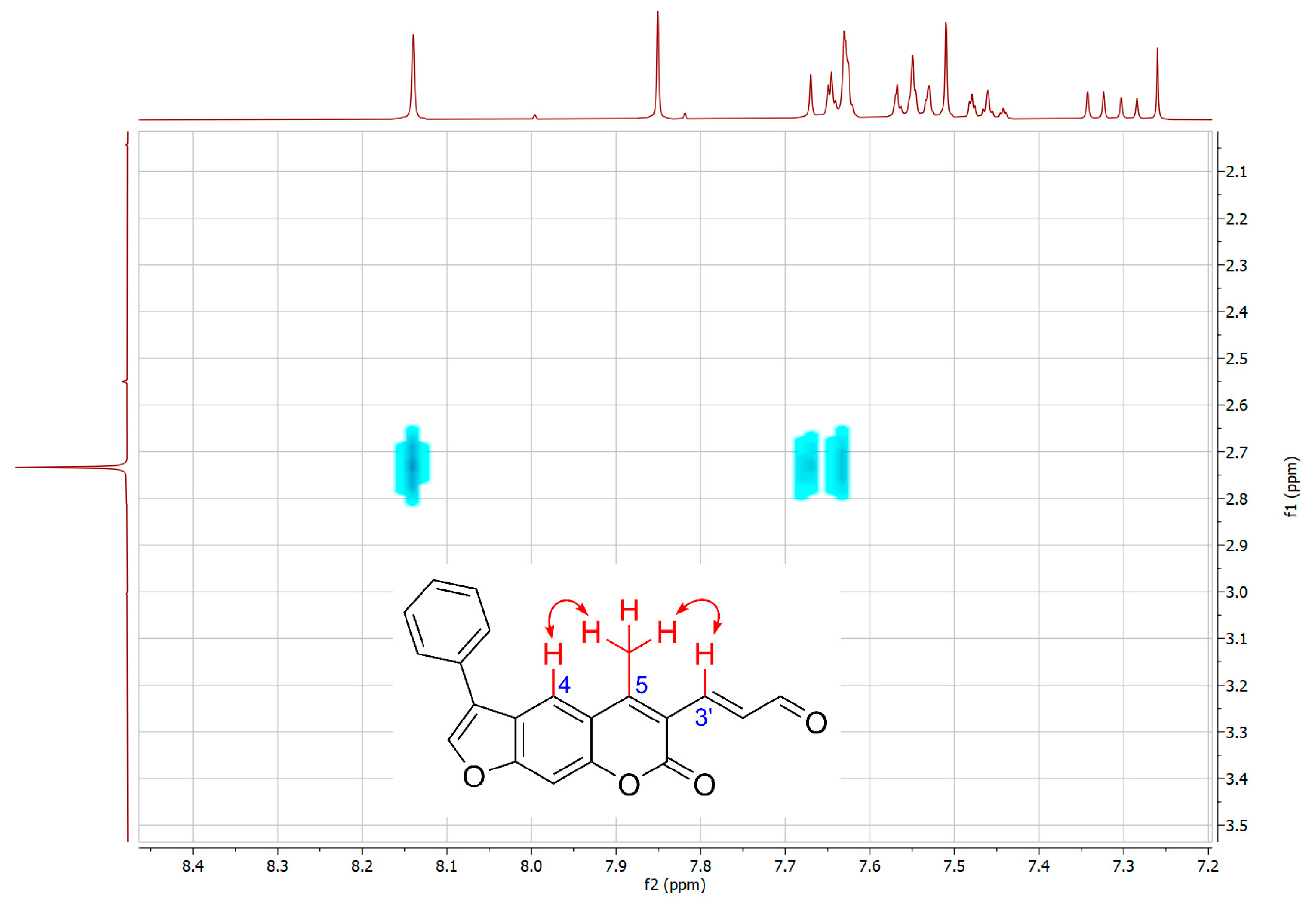
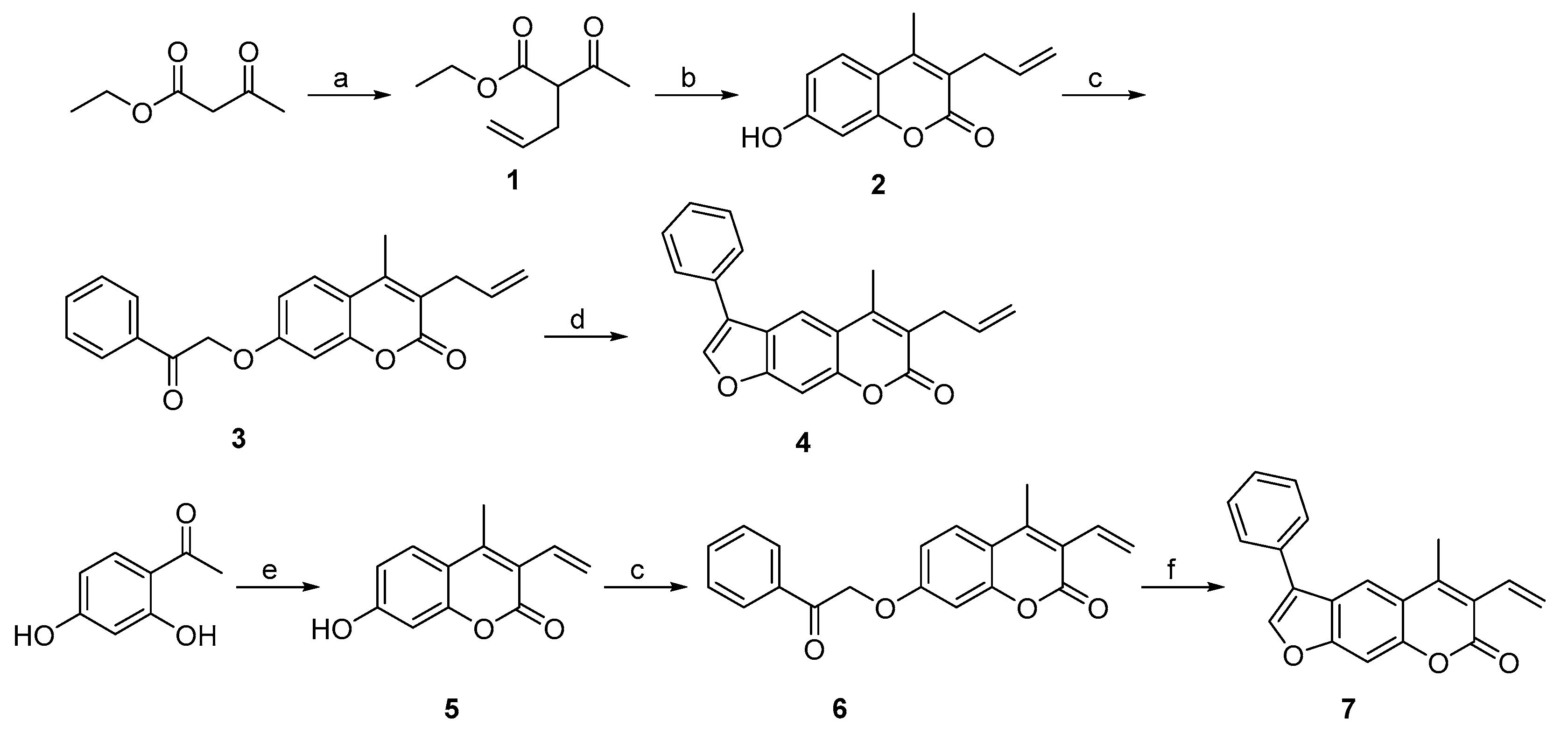
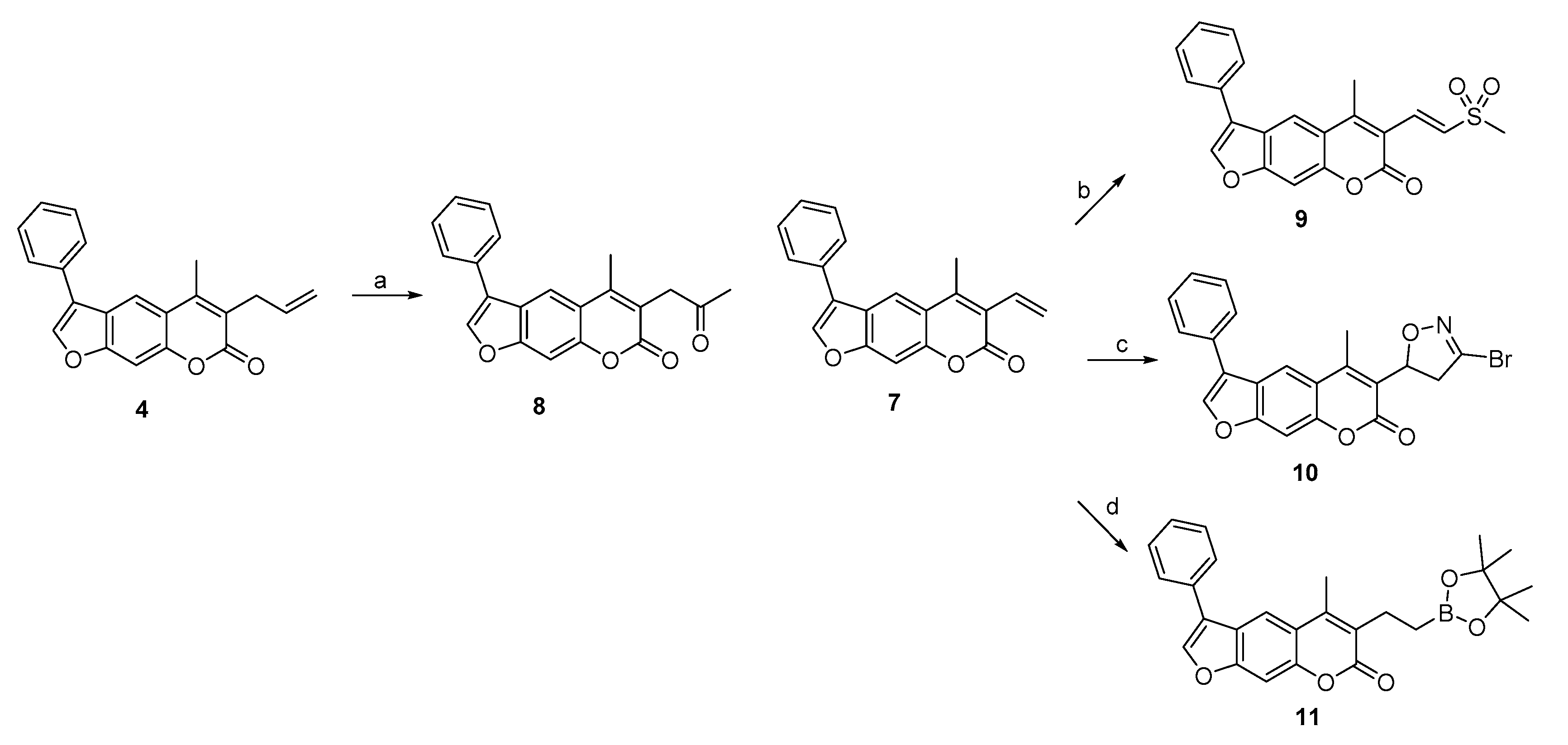
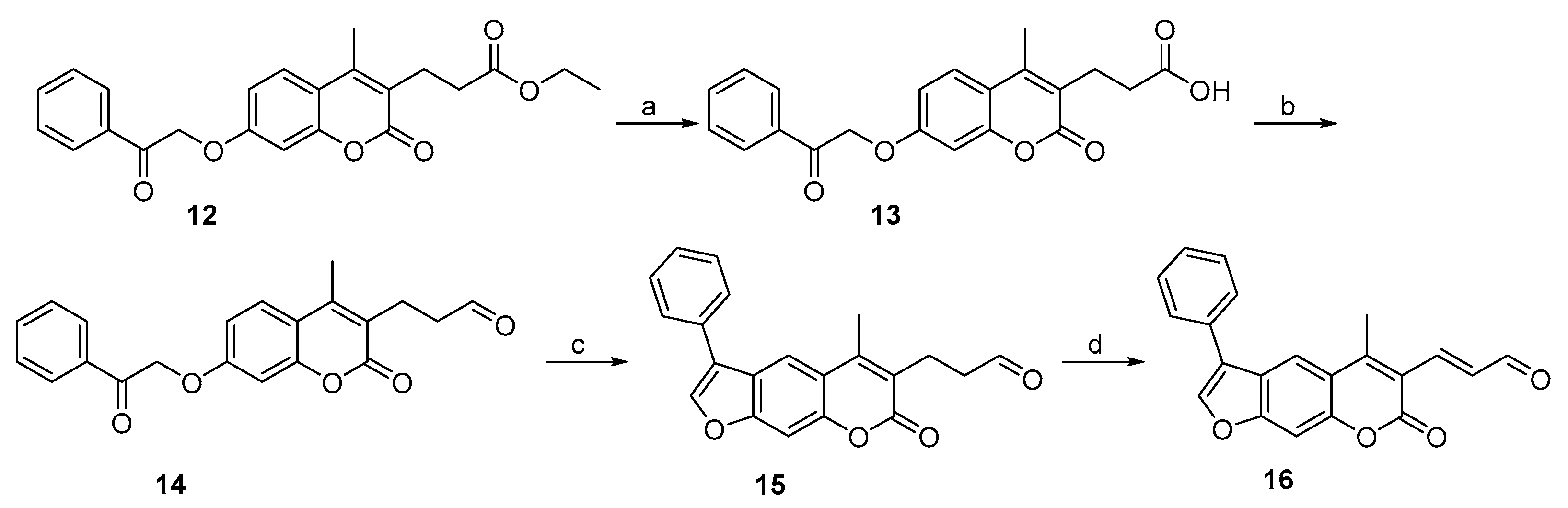
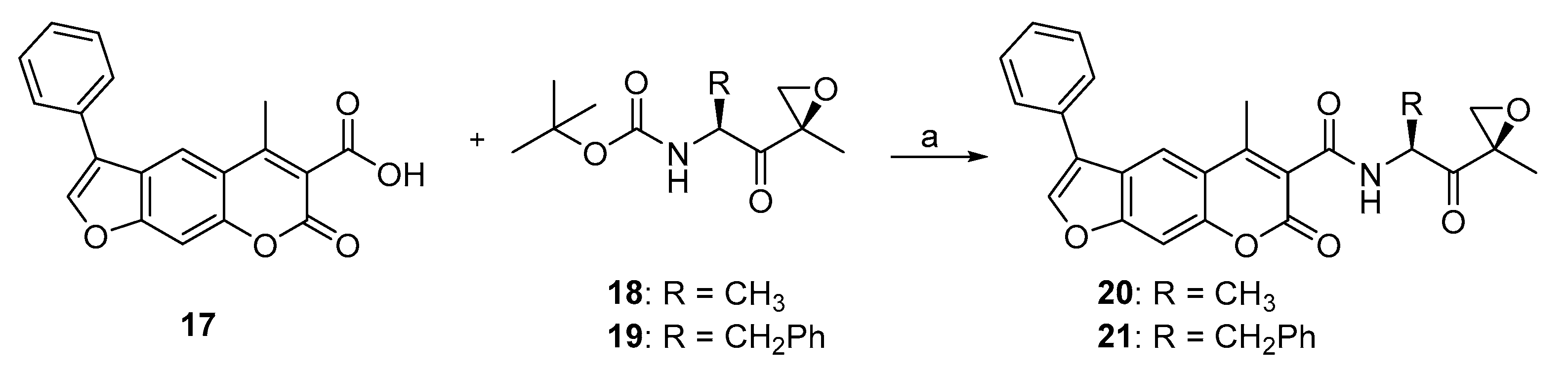
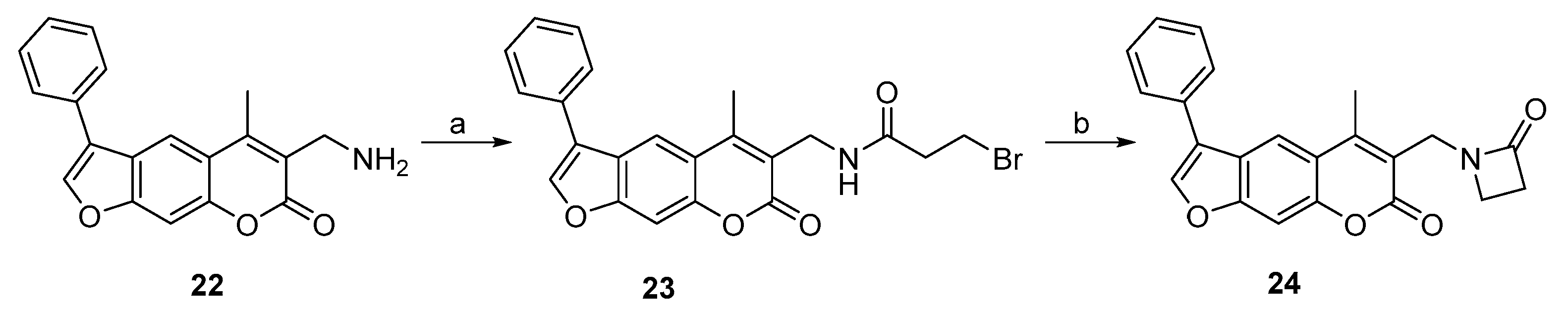
2.1. Syntheses of 3-Substituted Psoralens
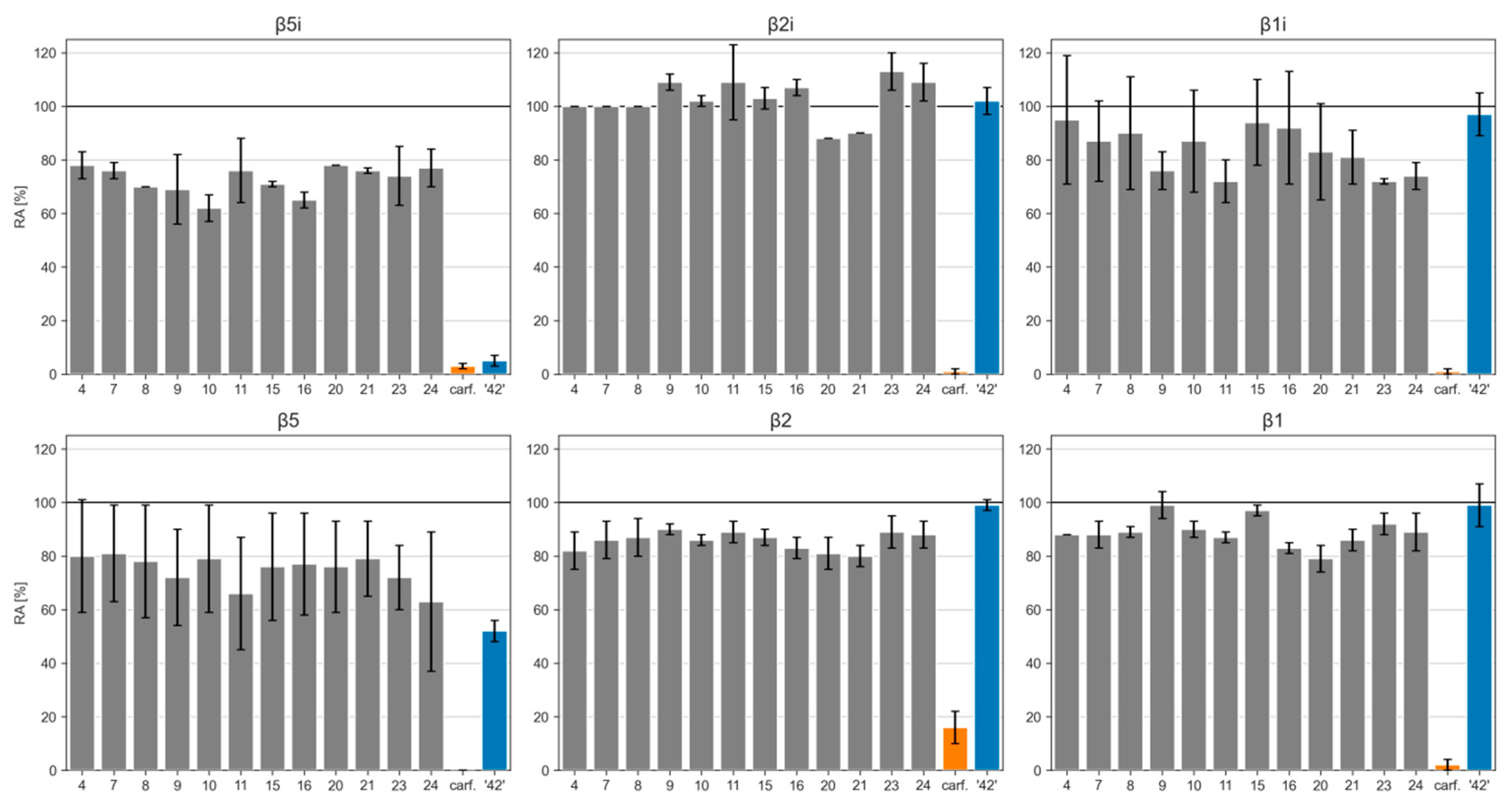
2.2. Biochemical Evaluation
3. Materials and Methods
3.1. General Chemistry Methods
3.2. Syntheses
3.3. Residual Activity Measurements
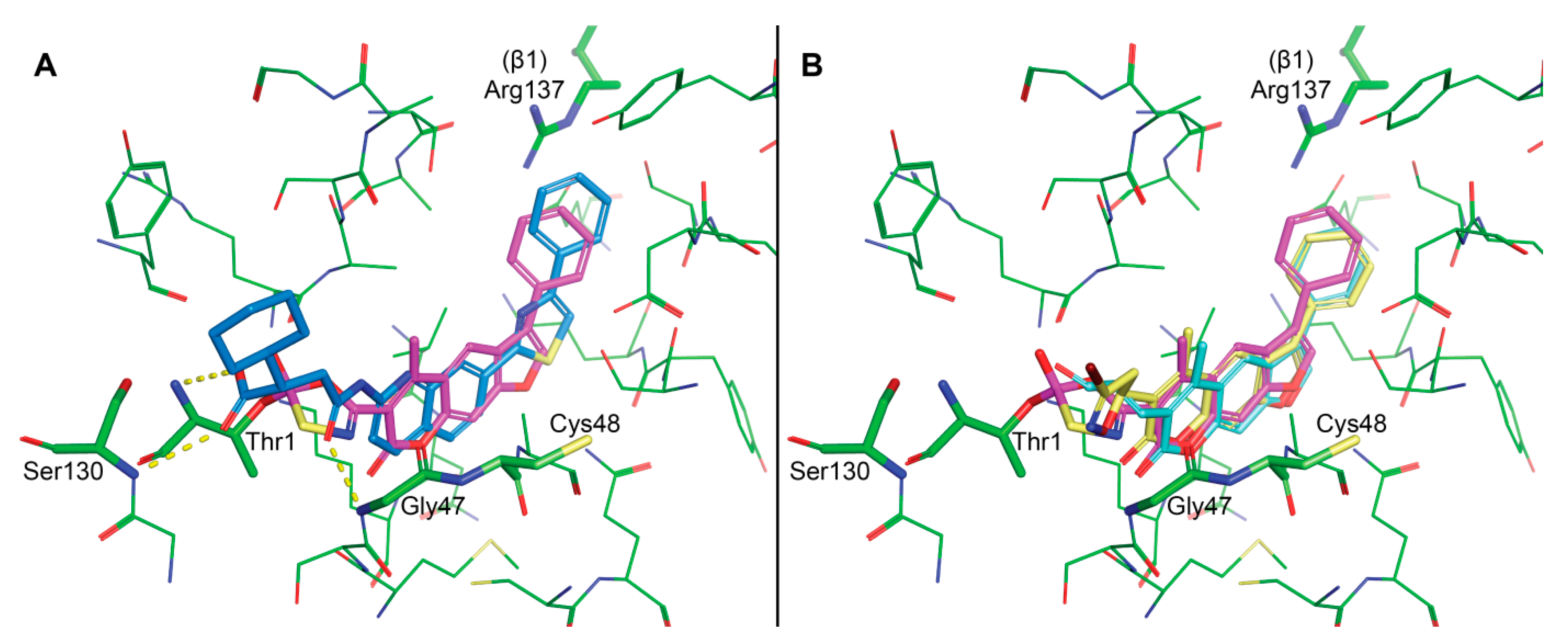
3.4. Molecular Modelling
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [Green Version]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- DeMartino, G.N.; Gillette, T.G. Proteasomes: Machines for all reasons. Cell 2007, 129, 659–662. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.A.; Goldberg, A.L. The logic of the 26S proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, A.L. Functions of the proteasome: From protein degradation and immune surveillance to cancer therapy. Biochem. Soc. Trans. 2007, 35, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Gallastegui, N.; Groll, M. The 26S proteasome: Assembly and function of a destructive machine. Trends Biochem. Sci. 2010, 35, 634–642. [Google Scholar] [CrossRef]
- Thibaudeau, T.A.; Smith, D.M. A practical review of proteasome pharmacology. Pharmacol. Rev. 2019, 71, 170–197. [Google Scholar] [CrossRef] [Green Version]
- Groll, M.; Ditzel, L.; Lowe, J.; Stock, D.; Bochtler, M.; Bartunikt, H.D.; Huber, R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 1997, 386, 463–471. [Google Scholar] [CrossRef]
- Arendt, C.S.; Hochstrasser, M. Identification of the Yeast 20S Proteasome Catalytic Centers and Subunit Interactions Required for Active-Site Formation. Proc. Natl. Acad. Sci. USA 1997, 94, 7156–7161. [Google Scholar] [CrossRef] [Green Version]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome structure and assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef]
- Murata, S.; Sasaki, K.; Kishimoto, T.; Niwa, S.-I.; Hayashi, H.; Takahama, Y.; Tanaka, K. Regulation of CD8+ T cell development by thymus-specific proteasomes. Science 2007, 316, 1349–1353. [Google Scholar] [CrossRef] [PubMed]
- Groettrup, M.; Kirk, C.J.; Basler, M. Proteasomes in immune cells: More than peptide producers? Nat. Rev. Immunol. 2010, 10, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K. Role of proteasomes modified by interferon-γ in antigen processing. J. Leukoc. Biol. 1994, 56, 571–575. [Google Scholar] [CrossRef]
- Aki, M.; Shimbara, N.; Takashina, M.; Akiyama, K.; Kagawa, S.; Tamura, T.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Interferon-γ induces different subunit organizations and functional diversity of proteasomes. J. Biochem. 1994, 115, 257–269. [Google Scholar] [CrossRef]
- Nandi, D.; Jiang, H.; Monaco, J.J. Identification of MECL-1 (LMP-10) as the third IFN-gamma-inducible proteasome subunit. J. Immunol. 1996, 156, 2361–2364. [Google Scholar]
- Basler, M.; Kirk, C.J.; Groettrup, M. The immunoproteasome in antigen processing and other immunological functions. Curr. Opin. Immunol. 2013, 25, 74–80. [Google Scholar] [CrossRef] [Green Version]
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, D.J.; Orlowski, R.Z. The immunoproteasome as a target in hematologic malignancies. Semin. Hematol. 2012, 49, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Ito, S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [Green Version]
- Verbrugge, E.E.; Scheper, R.J.; Lems, W.F.; de Gruijl, T.D.; Jansen, G. Proteasome inhibitors as experimental therapeutics of autoimmune diseases. Arthritis Res. Ther. 2015, 17, 17. [Google Scholar] [CrossRef] [Green Version]
- Basler, M.; Mundt, S.; Bitzer, A.; Schmidt, C.; Groettrup, M. The immunoproteasome: A novel drug target for autoimmune diseases. Clin. Exp. Rheumatol. 2015, 33, 74–79. [Google Scholar]
- Zhang, C.; Zhu, H.; Shao, J.; He, R.; Xi, J.; Zhuang, R.; Zhang, J. Immunoproteasome-selective inhibitors: The future of autoimmune diseases? Future Med. Chem. 2020, 12, 269–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limanaqi, F.; Biagioni, F.; Gaglione, A.; Busceti, C.L.; Fornai, F. A sentinel in the crosstalk between the nervous and immune system: The (immuno)-proteasome. Front. Immunol. 2019, 10, 628. [Google Scholar] [CrossRef] [Green Version]
- Huber, E.M.; Groll, M. Inhibitors for the immuno- and constitutive proteasome: Current and future trends in drug development. Angew. Chem. Int. Ed. 2012, 51, 8708–8720. [Google Scholar] [CrossRef]
- Cromm, P.M.; Crews, C.M. The proteasome in modern drug discovery: Second life of a highly valuable drug target. ACS Cent. Sci. 2017, 3, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Richy, N.; Sarraf, D.; Maréchal, X.; Janmamode, N.; Le Guével, R.; Genin, E.; Reboud-Ravaux, M.; Vidal, J. Structure-based design of human immuno- and constitutive proteasomes inhibitors. Eur. J. Med. Chem. 2018, 145, 570–587. [Google Scholar] [CrossRef]
- Sherman, D.J.; Li, J. Proteasome inhibitors: Harnessing proteostasis to combat disease. Molecules 2020, 25, 671. [Google Scholar] [CrossRef] [Green Version]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 7, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Johnson, H.W.B.; Lowe, E.; Anderl, J.L.; Fan, A.; Muchamuel, T.; Bowers, S.; Moebius, D.C.; Kirk, C.; McMinn, D.L. Required immunoproteasome subunit inhibition profile for anti-inflammatory efficacy and clinical candidate KZR-616 ((2S,3R)-N-((S)-3-(cyclopent-1-en-1-yl)-1-((R)-2-methyloxiran-2-yl)-1-oxopropan-2-yl)-3-hydroxy-3-(4-methoxyphenyl)-2-((S)-2-(2-morpholinoacetamido)propanamido)propenamide). J. Med. Chem. 2018, 61, 11127–11143. [Google Scholar] [CrossRef] [PubMed]
- Basler, M.; Lindstrom, M.M.; LaStant, J.J.; Bradshaw, J.M.; Owens, T.D.; Schmidt, C.; Meurits, E.; Tsu, C.; Overkleeft, H.S.; Kirk, C.J.; et al. Co-inhibition of immunoproteasome subunits LMP2 and LMP7 is required to block autoimmunity. EMBO Rep. 2018, 19, e46512. [Google Scholar] [CrossRef] [PubMed]
- Ettari, R.; Zappalà, M.; Grasso, S.; Musolino, C.; Innao, V.; Allegra, A. Immunoproteasome-selective and non-selective inhibitors: A promising approach for the treatment of multiple myeloma. Pharmacol. Ther. 2018, 182, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Zhuang, R.; Kong, L.; He, R.; Zhu, H.; Zhang, J. Immunoproteasome-selective inhibitors: An overview of recent developments as potential drugs for hematologic malignancies and autoimmune diseases. Eur. J. Med. Chem. 2019, 182, 111646. [Google Scholar] [CrossRef]
- Ladi, E.; Everett, C.; Stivala, C.E.; Daniels, B.E.; Durk, M.R.; Harris, S.F.; Huestis, M.P.; Purkey, H.E.; Staben, S.T.; Augustin, M.; et al. Design and evaluation of highly selective human immunoproteasome inhibitors reveal a compensatory process that preserves immune cell viability. J. Med. Chem. 2019, 62, 7032–7041. [Google Scholar] [CrossRef] [PubMed]
- Karreci, E.S.; Fan, H.; Uehara, M.; Mihali, A.B.; Singh, P.K.; Kurdi, A.T.; Solhjou, Z.; Riella, L.V.; Ghobrial, I.; Laragione, T.; et al. Brief treatment with a highly selective immunoproteasome inhibitor promotes long-term cardiac allograft acceptance in mice. Proc. Natl. Acad. Sci. USA 2016, 113, E8425–E8432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogorevc, E.; Schiffrer, E.S.; Sosič, I.; Gobec, S. A patent review of immunoproteasome inhibitors. Expert Opin. Ther. Pat. 2018, 28, 517–540. [Google Scholar] [CrossRef]
- Tan, C.R.C.; Abdul-Majeed, S.; Cael, B.; Barta, S.K. Clinical pharmacokinetics and pharmacodynamics of bortezomib. Clin. Pharmacokinet. 2019, 58, 157–168. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Z.; Fang, Y.; Jiang, J.; Zhao, F.; Wong, H.; Bennett, M.K.; Molineaux, C.J.; Kirk, C.J. Pharmacokinetics, pharmacodynamics, metabolism, distribution, and excretion of carfilzomib in rats. Drug Metab. Dispos. 2011, 39, 1873–1882. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Yang, J.; Kirk, C.; Fang, Y.; Alsina, M.; Badros, A.; Papadopoulos, K.; Wong, A.; Woo, T.; Bomba, D.; et al. Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab. Dispos. 2013, 41, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Sosič, I.; Gobec, M.; Brus, B.; Knez, D.; Živec, M.; Konc, J.; Lešnik, S.; Ogrizek, M.; Obreza, A.; Žigon, D.; et al. Nonpeptidic selective inhibitors of the chymotrypsin-like (β5i) subunit of the immunoproteasome. Angew. Chem. Int. Ed. 2016, 55, 5745–5748. [Google Scholar] [CrossRef]
- Schiffrer, E.S.; Sosič, I.; Šterman, A.; Mravljak, J.; Raščan, I.M.; Gobec, S.; Gobec, M. A focused structure-activity relationship study of psoralen-based immunoproteasome inhibitors. Med. Chem. Commun. 2019, 10, 1958–1965. [Google Scholar] [CrossRef]
- Fan, H.; Angelo, N.G.; Warren, J.D.; Nathan, C.F.; Lin, G. Oxathiazolones selectively inhibit the human immunoproteasome over the constitutive proteasome. ACS Med. Chem. Lett. 2014, 5, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Kasam, V.; Lee, N.-R.; Kim, K.-B.; Zhan, C.-G. Selective immunoproteasome inhibitors with non-peptide scaffolds identified from structure-based virtual screening. Bioorg. Med. Chem. Lett. 2014, 24, 3614–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Baur, R.; Le Chapelain, C.; Dubiella, C.; Heinemeyer, W.; Huber, E.M.; Groll, M. Structural elucidation of a nonpeptidic inhibitor specific for the human immunoproteasome. ChemBioChem 2017, 18, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Scarpino, A.; Bajusz, D.; Proj, M.; Gobec, M.; Sosič, I.; Gobec, S.; Ferenczy, G.G.; Keseru, G.M. Discovery of immunoproteasome inhibitors using large-scale covalent virtual screening. Molecules 2019, 24, 2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.; Tamborini, L.; Cullia, G.; Conti, P.; De Micheli, C. Inspired by nature: The 3-halo-4,5-dihydroisoxazole moiety as a novel molecular warhead for the design of covalent inhibitors. ChemMedChem 2016, 11, 10–14. [Google Scholar] [CrossRef]
- Huber, E.M.; de Bruin, G.; Heinemeyer, W.; Soriano, G.P.; Overkleeft, H.S.; Groll, M. Systematic analyses of substrate preferences of 20S proteasomes using peptidic epoxyketone inhibitors. J. Am. Chem. Soc. 2015, 137, 7835–7842. [Google Scholar] [CrossRef]
- Zhou, H.J.; Aujay, M.A.; Bennett, M.K.; Dajee, M.; Demo, S.D.; Fang, Y.; Ho, M.N.; Jiang, J.; Kirk, C.J.; Laidig, G.J.; et al. Design and synthesis of an orally bioavailable and selective peptide epoxyketone proteasome inhibitor (PR-047). J. Med. Chem. 2009, 52, 3028–3038. [Google Scholar] [CrossRef]
- Chaudhari, D.A.; Fernandes, R.A. Hypervalent iodine as a terminal oxidant in wacker-type oxidation of terminal olefins to methyl ketones. J. Org. Chem. 2016, 81, 2113–2121. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking covalent inhibitors: A parameter free approach to pose prediction and scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | β5i (RA [%]) 1 | β2i (RA [%]) 1 | β1i (RA [%]) 1 | β5 (RA [%]) 1 | β2 (RA [%]) 1 | β1 (RA [%]) 1 |
|---|---|---|---|---|---|---|
| 4 | 78 ± 5 | 100 ± 0 | 95 ± 24 | 80 ± 21 | 82 ± 7 | 88 ± 0 |
| 7 | 76 ± 3 | 100 ± 0 | 87 ± 15 | 81 ± 18 | 86 ± 7 | 88 ± 5 |
| 8 | 70 ± 0 | 100 ± 0 | 90 ± 21 | 78 ± 21 | 87 ± 7 | 89 ± 2 |
| 9 | 69 ± 13 | 109 ± 3 | 76 ± 7 | 72 ± 18 | 90 ± 2 | 99 ± 5 |
| 10 | 62 ± 5 | 102 ± 2 | 87 ± 19 | 79 ± 20 | 86 ± 2 | 90 ± 3 |
| 11 | 76 ± 12 | 109 ± 14 | 72 ± 8 | 66 ± 21 | 89 ± 4 | 87 ± 2 |
| 15 | 71 ± 1 | 103 ± 4 | 94 ± 16 | 76 ± 20 | 87 ± 3 | 97 ± 2 |
| 16 | 65 ± 3 | 107 ± 3 | 92 ± 21 | 77 ± 19 | 83 ± 4 | 83 ± 2 |
| 20 | 78 ± 0 | 88 ± 0 | 83 ± 18 | 76 ± 17 | 81 ± 6 | 79 ± 5 |
| 21 | 76 ± 1 | 90 ± 0 | 81 ± 10 | 79 ± 14 | 80 ± 4 | 86 ± 4 |
| 23 | 74 ± 11 | 113 ± 7 | 72 ± 1 | 72 ± 12 | 89 ± 6 | 92 ± 4 |
| 24 | 77 ± 7 | 109 ± 7 | 74 ± 5 | 63 ± 26 | 88 ± 5 | 89 ± 7 |
| carf. | 3 ± 1 | 1 ± 1 | 1 ± 1 | 0 ± 0 | 16 ± 6 | 2 ± 2 |
| ‘42’ | 5 ± 2 | 102 ± 5 | 97 ± 8 | 52 ± 4 | 99 ± 2 | 99 ± 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiffrer, E.S.; Proj, M.; Gobec, M.; Rejc, L.; Šterman, A.; Mravljak, J.; Gobec, S.; Sosič, I. Synthesis and Biochemical Evaluation of Warhead-Decorated Psoralens as (Immuno)Proteasome Inhibitors. Molecules 2021, 26, 356. https://doi.org/10.3390/molecules26020356
Schiffrer ES, Proj M, Gobec M, Rejc L, Šterman A, Mravljak J, Gobec S, Sosič I. Synthesis and Biochemical Evaluation of Warhead-Decorated Psoralens as (Immuno)Proteasome Inhibitors. Molecules. 2021; 26(2):356. https://doi.org/10.3390/molecules26020356
Chicago/Turabian StyleSchiffrer, Eva Shannon, Matic Proj, Martina Gobec, Luka Rejc, Andrej Šterman, Janez Mravljak, Stanislav Gobec, and Izidor Sosič. 2021. "Synthesis and Biochemical Evaluation of Warhead-Decorated Psoralens as (Immuno)Proteasome Inhibitors" Molecules 26, no. 2: 356. https://doi.org/10.3390/molecules26020356
APA StyleSchiffrer, E. S., Proj, M., Gobec, M., Rejc, L., Šterman, A., Mravljak, J., Gobec, S., & Sosič, I. (2021). Synthesis and Biochemical Evaluation of Warhead-Decorated Psoralens as (Immuno)Proteasome Inhibitors. Molecules, 26(2), 356. https://doi.org/10.3390/molecules26020356