
Development of Microchip Isotachophoresis Coupled with Ion Mobility Spectrometry and Evaluation of Its Potential for the Analysis of Food, Biological and Pharmaceutical Samples

 , ,
, , 
Abstract
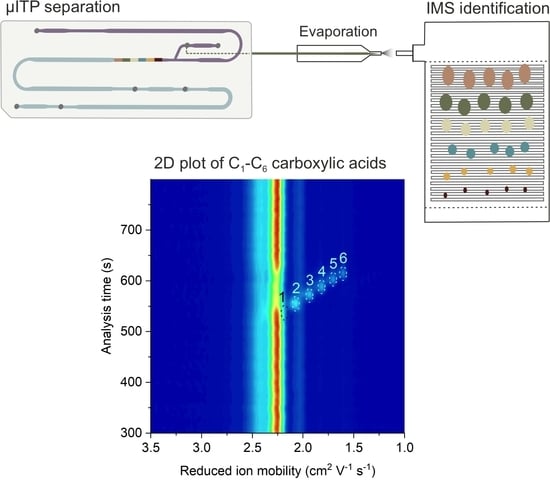
:
1. Introduction
2. Results and Discussion
2.1. Optimization of µITP Separation Conditions
2.2. Optimization of µITP-IMS Transfer Parameters
2.3. µITP-IMS Method Performance Parameters
2.4. Analysis of Food, Biological and Pharmaceutical Samples
3. Materials and Methods
3.1. Chemicals and Solutions
3.2. Food, Biological and Pharmaceutical Samples
3.3. µITP-IMS Instrumentation
3.4. Microchip Maintenance and Measurement Procedure
3.5. Data Processing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Robles, A.; Fabjanowicz, M.; Chmiel, T.; Płotka-Wasylka, J. Determination and identification of organic acids in wine samples. Problems and challenges. TrAC Trends Anal. Chem. 2019, 120, 115630. [Google Scholar] [CrossRef]
- The European Commission. Commission Regulation (EU) No 231/2012 of 9 March 2012 laying down specifications for food additives listed in Annexes II and III to Regulation (EC) No 1333/2008 of the European Parliament and of the Council. Off. J. Eur. Union 2012, L83, 1–295. [Google Scholar]
- Van Balen, F.A.M.; Smit, W.M.; Zuithoff, N.P.A.; Verheij, T.J.M. Clinical efficacy of three common treatments in acute otitis externa in primary care: Randomised controlled trial. Br. Med. J. 2003, 327, 1201–1203. [Google Scholar] [CrossRef] [Green Version]
- Baena, B.; Cifuentes, A.; Barbas, C. Analysis of carboxylic acids in biological fluids by capillary electrophoresis. Electrophoresis 2005, 26, 2622–2636. [Google Scholar] [CrossRef]
- Qiu, X.; Zhang, Y.; Zhou, Y.; Li, G.H.; Feng, X.S. Progress in pretreatment and analysis of organic Acids: An update since 2010. Food Chem. 2021, 360, 129977. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; Ropartz, D.; García-Campaña, A.M.; Rogniaux, H.; Dervilly-Pinel, G.; Le Bizec, B. Ion mobility spectrometry in food analysis: Principles, current applications and future trends. Molecules 2019, 24, 2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gałuszka, A.; Migaszewski, Z.; Namieśnik, J. The 12 principles of green analytical chemistry and the SIGNIFICANCE mnemonic of green analytical practices. TrAC Trends Anal. Chem. 2013, 50, 78–84. [Google Scholar] [CrossRef]
- Ferey, L.; Delaunay, N. Food Analysis on Electrophoretic Microchips. Sep. Purif. Rev. 2016, 45, 193–226. [Google Scholar] [CrossRef]
- Nuchtavorn, N.; Suntornsuk, W.; Lunte, S.M.; Suntornsuk, L. Recent applications of microchip electrophoresis to biomedical analysis. J. Pharm. Biomed. Anal. 2015, 113, 72–96. [Google Scholar] [CrossRef]
- Chouinard, C.D.; Wei, M.S.; Beekman, C.R.; Kemperman, R.H.J.; Yost, R.A. Ion mobility in clinical analysis: Current progress and future perspectives. Clin. Chem. 2016, 62, 124–133. [Google Scholar] [CrossRef] [Green Version]
- Márquez-Sillero, I.; Aguilera-Herrador, E.; Cárdenas, S.; Valcárcel, M. Ion-mobility spectrometry for environmental analysis. TrAC Trends Anal. Chem. 2011, 30, 677–690. [Google Scholar] [CrossRef]
- Zheng, X.; Wojcik, R.; Zhang, X.; Ibrahim, Y.M.; Burnum-Johnson, K.E.; Orton, D.J.; Monroe, M.E.; Moore, R.J.; Smith, R.D.; Baker, E.S. Coupling front-end separations, ion mobility spectrometry, and mass spectrometry for enhanced multidimensional biological and environmental analyses. Annu. Rev. Anal. Chem. 2017, 10, 71–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Purves, R.W.; Richards, J.C. Coupling capillary electrophoresis and high-field asymmetric waveform ion mobility spectrometry mass spectrometry for the analysis of complex lipopolysaccharides. Anal. Chem. 2004, 76, 4676–4683. [Google Scholar] [CrossRef]
- Revercomb, H.E.; Mason, E.A. Theory of Plasma Chromatography/Gaseous Electrophoresis. A Review. Anal. Chem. 1975, 47, 970–983. [Google Scholar] [CrossRef]
- Jiang, X.; Svensmark, B.; Deng, L. Coupling capillary electrophoresis and ion mobility spectrometry via electrospray interface: A preliminary study. Adv. Mater. Res. 2011, 160–162, 1531–1534. [Google Scholar] [CrossRef]
- Shumate, C.B.; Hill, H.H., Jr. Coronaspray Nebulization and Ionization of Liquid Samples for Ion Mobility Spectrometry. Anal. Chem. 1989, 61, 601–606. [Google Scholar] [CrossRef]
- Hallen, R.W.; Shumate, C.B.; Siems, W.F.; Tsuda, T.; Hill, H.H., Jr. Preliminary investigation of ion mobility spectrometry after capillary electrophoretic introduction. J. Chromatogr. A 1989, 480, 233–245. [Google Scholar] [CrossRef]
- Masár, M.; Hradski, J.; Nováková, M.; Szucs, R.; Sabo, M.; Matejčík, S. Online coupling of microchip electrophoresis with ion mobility spectrometry for direct analysis of complex liquid samples. Sens. Actuators B Chem. 2020, 302, 127183. [Google Scholar] [CrossRef]
- Hartner, N.T.; Raddatz, C.R.; Thoben, C.; Piendl, S.K.; Zimmermann, S.; Belder, D. On-line coupling of chip-electrochromatography and ion mobility spectrometry. Anal. Chem. 2020, 92, 15129–15136. [Google Scholar] [CrossRef] [PubMed]
- Hradski, J.; Chorváthová, M.D.; Bodor, R.; Sabo, M.; Matejčík, Š.; Masár, M. Quantitative aspects of microchip isotachophoresis for high precision determination of main components in pharmaceuticals. Anal. Bioanal. Chem. 2016, 408, 8669–8679. [Google Scholar] [CrossRef]
- Masár, M.; Troška, P.; Hradski, J.; Talian, I. Microchip isotachophoresis coupled to surface-enhanced Raman spectroscopy for pharmaceutical analysis. Microchim. Acta 2020, 187, 448. [Google Scholar] [CrossRef]
- Malý, M.; Boublík, M.; Pocrnić, M.; Ansorge, M.; Lorinčíková, K.; Svobodová, J.; Hruška, V.; Dubský, P.; Gaš, B. Determination of thermodynamic acidity constants and limiting ionic mobilities of weak electrolytes by capillary electrophoresis using a new free software AnglerFish. Electrophoresis 2020, 41, 493–501. [Google Scholar] [CrossRef]
- Tao, L.; Han, J.; Tao, F.M. Correlations and predictions of carboxylic acid pKa values using intermolecular structure and properties of hydrogen-bonded complexes. J. Phys. Chem. A 2008, 112, 775–782. [Google Scholar] [CrossRef]
- Kaniansky, D.; Masár, M.; Bodor, R.; Žúborová, M.; Ölvecká, M.; Jöhnck, M.; Stanislawski, B. Electrophoretic separations on chips with hydrodynamically closed separation systems. Electrophoresis 2003, 24, 2208–2227. [Google Scholar] [CrossRef]
- Sabo, M.; Malásková, M.; Harmathová, O.; Hradski, J.; Masár, M.; Radjenovic, B.; Matejčík, S. Direct Liquid Sampling for Corona Discharge Ion Mobility Spectrometry. Anal. Chem. 2015, 87, 7389–7394. [Google Scholar] [CrossRef] [PubMed]
- Sabo, M.; Matúška, J.; Matejčík, S. Specific O2− generation in corona discharge for ion mobility spectrometry. Talanta 2011, 85, 400–405. [Google Scholar] [CrossRef]
- Gao, H.; Niu, W.; Hong, Y.; Xu, B.; Shen, C.; Huang, C.; Jiang, H.; Chu, Y. Negative photoionization chloride ion attachment ion mobility spectrometry for the detection of organic acids. RSC Adv. 2014, 4, 63977–63984. [Google Scholar] [CrossRef]
- Sengun, I.Y.; Kilic, G.; Ozturk, B. Screening physicochemical, microbiological and bioactive properties of fruit vinegars produced from various raw materials. Food Sci. Biotechnol. 2020, 29, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Park, J.N.; Fukumoto, Y.; Fujita, E.; Tanaka, T.; Washio, T.; Otsuka, S.; Shimizu, T.; Watanabe, K.; Abe, H. Chemical Composition of Fish Sauces Produced in Southeast and East Asian Countries. J. Food Compos. Anal. 2001, 14, 113–125. [Google Scholar] [CrossRef]
- Park, Y.D.; Jang, J.H.; Oh, Y.J.; Kwon, H.J. Analyses of organic acids and inorganic anions and their relationship in human saliva before and after glucose intake. Arch. Oral Biol. 2014, 59, 1–11. [Google Scholar] [CrossRef]
- Troška, P.; Chudoba, R.; Danč, L.; Bodor, R.; Horčičiak, M.; Tesařová, E.; Masár, M. Determination of nitrite and nitrate in cerebrospinal fluid by microchip electrophoresis with microsolid phase extraction pre-treatment. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 930, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Eiceman, G.A.; Nazarov, E.G.; Stone, J.A. Chemical standards in ion mobility spectrometry. Anal. Chim. Acta 2003, 493, 185–194. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | LOD [mg L−1] | LOQ [mg L−1] | Equation of Regression Line | Correlation Coefficient | Accuracy [%] a | K0 [cm2 V−1 s−1] (RSD [%]) a |
|---|---|---|---|---|---|---|
| Acetic acid | 0.13 | 0.39 | y = 0.561x + 0.178 | 0.999 | 99.7–101.5 | 2.07 (0.23) |
| Propionic acid | 0.15 | 0.46 | y = 0.518x + 0.096 | 0.999 | 97.5–101.1 | 1.94 (0.22) |
| Butyric acid | 0.19 | 0.58 | y = 0.428x + 0.089 | 0.999 | 99.3–101.7 | 1.82 (0.27) |
| Valeric acid | 0.23 | 0.70 | y = 0.375x + 0.023 | 0.999 | 99.2–100.7 | 1.70 (0.30) |
| Caproic acid | 0.31 | 0.93 | y = 0.278x + 0.053 | 0.999 | 98.5–102.4 | 1.60 (0.29) |
| Analyte | Concentration [mg L−1] | Accuracy [%] a | K0 [cm2 V−1 s−1] (RSD [%]) a |
|---|---|---|---|
| Acetic acid | 79,221.5 b; 212.3 c; 901.2 d; 130.2 e; 50,124.5 f | 92.5–108.3 | 2.06 (0.40) |
| Propionic acid | 44.7 e | 93.0–108.0 | 1.93 (0.29) |
| Butyric acid | n.d. | 100.9–108.7 | 1.81 (0.50) |
| Valeric acid | n.d. | 94.5–107.7 | 1.69 (0.58) |
| Caproic acid | n.d. | 93.9–104.2 | 1.60 (0.31) |
| Instrument | Parameter | Setting |
|---|---|---|
| MCE analyzer | operating mode | ITP–anionic |
| driving current | 20 (10 at detection point) µA | |
| effective separation path (length × width × depth) | 59 × 0.2–0.5 × 0.14–0.2 mm | |
| Evaporation unit | flow rate | 30 µL min−1 |
| temperature of droplet stream | 130 °C | |
| IMS analyzer | operating mode | negative |
| sample gas flow rate | 100 mL min−1 | |
| shutter grid pulse width | 100 µs | |
| drift gas flow rate | 600 mL min−1 | |
| drift field intensity | 565 V cm−1 | |
| drift tube temperature | 100 °C | |
| operating pressure | 600 mbar | |
| acquisition time | 130 ms |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hradski, J.; Ďuriš, M.; Szucs, R.; Moravský, L.; Matejčík, Š.; Masár, M. Development of Microchip Isotachophoresis Coupled with Ion Mobility Spectrometry and Evaluation of Its Potential for the Analysis of Food, Biological and Pharmaceutical Samples. Molecules 2021, 26, 6094. https://doi.org/10.3390/molecules26206094
Hradski J, Ďuriš M, Szucs R, Moravský L, Matejčík Š, Masár M. Development of Microchip Isotachophoresis Coupled with Ion Mobility Spectrometry and Evaluation of Its Potential for the Analysis of Food, Biological and Pharmaceutical Samples. Molecules. 2021; 26(20):6094. https://doi.org/10.3390/molecules26206094
Chicago/Turabian StyleHradski, Jasna, Marta Ďuriš, Roman Szucs, Ladislav Moravský, Štefan Matejčík, and Marián Masár. 2021. "Development of Microchip Isotachophoresis Coupled with Ion Mobility Spectrometry and Evaluation of Its Potential for the Analysis of Food, Biological and Pharmaceutical Samples" Molecules 26, no. 20: 6094. https://doi.org/10.3390/molecules26206094
APA StyleHradski, J., Ďuriš, M., Szucs, R., Moravský, L., Matejčík, Š., & Masár, M. (2021). Development of Microchip Isotachophoresis Coupled with Ion Mobility Spectrometry and Evaluation of Its Potential for the Analysis of Food, Biological and Pharmaceutical Samples. Molecules, 26(20), 6094. https://doi.org/10.3390/molecules26206094