
Microwave Heating Promotes the S-Alkylation of Aziridine Catalyzed by Molecular Sieves: A Post-Synthetic Approach to Lanthionine-Containing Peptides



Abstract
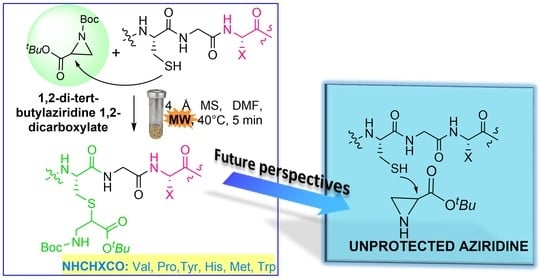
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Peptide Synthesis
3.2. General Procedure for the MW-Assisted Peptide S-Alkylation (Compound 6a)
Compound 1a strategy A. AcLanGlyValAlaNH2
Compound 2a strategy A. AcLanGlyProAlaNH2
Compound 3a strategy A. AcLanGlyTyrAlaNH2
Compound 4a strategy A. AcLanGlyHisAlaNH2
Compound 5a strategy A. AcLanGlyMetValAlaNH2
Compound 6a (Strategy A). AcGlyTrpLanHisValAlaNH2
Compound 1d 1H-NMR
Compound 6a (Strategy B). AcGlyTrpLanHisValAlaNH2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hata, Y.; Watanabe, M. Reaction of aziridinecarylic acids with thiols in aqueous solution. the formation of β-amino acid. Tetrahedron 1987, 43, 3881–3888. [Google Scholar] [CrossRef]
- Luginina, J.; Turks, M. Regioselective ring opening of NH-aziridines with sulfur nucleophiles in liquid SO2. Synlett 2017, 28, 939–943. [Google Scholar] [CrossRef]
- Pankova, A.S.; Kuznetsov, M.A. Recent Advances in the Chemistry of 2-Acylaziridines. Synthesis 2017, 49, 5093–5104. [Google Scholar] [CrossRef]
- Akhtar, R.; Naqvi, S.A.R.; Zahoor, A.F.; Saleem, S. Nucleophilic ring opening reactions of aziridines. Mol. Divers. 2018, 22, 447–501. [Google Scholar] [CrossRef]
- Denoël, T.; Lemaire, C.; Luxen, A. Progress in Lanthionine and Protected Lanthionine Synthesis. Chem. Eur. J. 2018, 24, 15421–15441. [Google Scholar] [CrossRef]
- Verdoliva, V.; Saviano, M.; De Luca, S. Zeolites as Acid/Basic Solid Catalysts: Recent Synthetic Developments. Catalysts 2019, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Calce, E.; Leone, M.; Monfregola, L.; De Luca, S. Chemical modifications of peptide sequences via S-alkylation reaction. Org. Lett. 2013, 15, 5354–5357. [Google Scholar] [CrossRef]
- Calce, E.; Leone, M.; Mercurio, F.A.; Monfregola, L.; DeLuca, S. Solid-Phase S-Alkylation Promoted by Molecular Sieves. Org. Lett. 2015, 17, 5646–5649. [Google Scholar] [CrossRef]
- Calce, E.; De Luca, S. The Cysteine S-Alkylation Reaction as a Synthetic Method to Covalently Modify Peptide Sequences. Chem. Eur. J. 2016, 22, 224–233. [Google Scholar] [CrossRef]
- Monfregola, L.; De Luca, S. Synthetic strategy for side chain mono-N-alkylation of Fmoc-amino acids promoted by molecular sieves. Amino Acids 2011, 41, 981–990. [Google Scholar] [CrossRef]
- Calce, E.; De Luca, L. Microwave heating in peptide side chain modification via cysteine alkylation. Amino Acids 2016, 48, 2267–2271. [Google Scholar] [CrossRef]
- Jain, A.K.; Singla, R.K. An overview of Microwave assisted technique: Green Synthesis. Webmed Cent. Pharm. Sci. 2011, 2, WMC002251. [Google Scholar] [CrossRef]
- Rosana, M.R.; Tao, Y.; Stiegman, A.E.; Dudley, G.B. On the rational design of microwave-actuated organic reactions. Chem. Sci. 2012, 3, 1240–1244. [Google Scholar] [CrossRef]
- Ravichandran, S.; Karthikeyan, E. Microwave Synthesis—A Potential Tool for Green Chemistry. Int. J. Chem. Tech. Res. 2011, 3, 466–470. [Google Scholar]
- Kappe, C.O.; Pieber, B.; Dallinger, D. Microwave Effects in Organic Synthesis: Myth or Reality? Angew. Chem. Int. Ed. 2013, 52, 1088–1094. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Rodríguez, A.M.; Prieto, P.; de la Hoz, A.; Diaz-Ortiz, A.; Martín, D.R.; Gracía, J.I. Influence of Polarity and Activation Energy in Microwave–Assisted Organic Synthesis (MAOS). ChemistryOpen 2015, 4, 308–317. [Google Scholar] [CrossRef] [Green Version]
- De Luca, S.; Digilio, G.; Verdoliva, V.; Saviano, M.; Menchise, V.; Tovillas, P.; Jimeénez-Oseés, G.; Peregrina, J.M. A Late-Stage Synthetic Approach to Lanthionine-Containing Peptides via S-Alkylation on Cyclic Sulfamidates Promoted by Molecular Sieves. Org. Lett. 2018, 20, 7478–7482. [Google Scholar] [CrossRef]
- De Luca, S.; Digilio, G.; Verdoliva, V.; Tovillas, P.; Jimeénez-Oseés, G.; Peregrina, J.M. Lanthionine Peptides by S-Alkylation with Substituted Cyclic Sulfamidates Promoted by Activated Molecular Sieves: Effects of the Sulfamidate Structure on the Yield. J. Org. Chem. 2019, 84, 14957–14964. [Google Scholar] [CrossRef]
- Liu, W.; Chan, A.S.; Liu, H.; Cochrane, S.A.; Vederas, J.C. Solid Supported Chemical Syntheses of Both Components of the Lantibiotic Lacticin 3147. J. Am. Chem. Soc. 2011, 133, 14216–14219. [Google Scholar] [CrossRef]
- Li, Z.; Gentry, Z.; Murphy, B.; VanNieuwenhze, M.S. Scalable Synthesis of Orthogonally Protected β-Methyllanthionines by Indium(III)-Mediated Ring Opening of Aziridines. Org. Let. 2019, 21, 2200–2203. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Peptide | Yield—MW b (%) | Yield—SC c (%) |
|---|---|---|---|
| 1a | AcLanGlyValAlaNH2 | 25 | - |
| 2a | AcLanGlyProAlaNH2 | 20 | - |
| 3a | AcLanGlyTyrAlaNH2 | 30 | - |
| 4a | AcLanGlyHisAlaNH2 | 25 | - |
| 5a | AcLanGlyMetValAlaNH2 | 20 | - |
| 6a | AcGlyTrpLanHisValAlaNH2 | 25 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdoliva, V.; Digilio, G.; Saviano, M.; De Luca, S. Microwave Heating Promotes the S-Alkylation of Aziridine Catalyzed by Molecular Sieves: A Post-Synthetic Approach to Lanthionine-Containing Peptides. Molecules 2021, 26, 6135. https://doi.org/10.3390/molecules26206135
Verdoliva V, Digilio G, Saviano M, De Luca S. Microwave Heating Promotes the S-Alkylation of Aziridine Catalyzed by Molecular Sieves: A Post-Synthetic Approach to Lanthionine-Containing Peptides. Molecules. 2021; 26(20):6135. https://doi.org/10.3390/molecules26206135
Chicago/Turabian StyleVerdoliva, Valentina, Giuseppe Digilio, Michele Saviano, and Stefania De Luca. 2021. "Microwave Heating Promotes the S-Alkylation of Aziridine Catalyzed by Molecular Sieves: A Post-Synthetic Approach to Lanthionine-Containing Peptides" Molecules 26, no. 20: 6135. https://doi.org/10.3390/molecules26206135
APA StyleVerdoliva, V., Digilio, G., Saviano, M., & De Luca, S. (2021). Microwave Heating Promotes the S-Alkylation of Aziridine Catalyzed by Molecular Sieves: A Post-Synthetic Approach to Lanthionine-Containing Peptides. Molecules, 26(20), 6135. https://doi.org/10.3390/molecules26206135