
A Neoglycoprotein-Immobilized Fluorescent Magnetic Bead Suspension Multiplex Array for Galectin-Binding Studies


Abstract
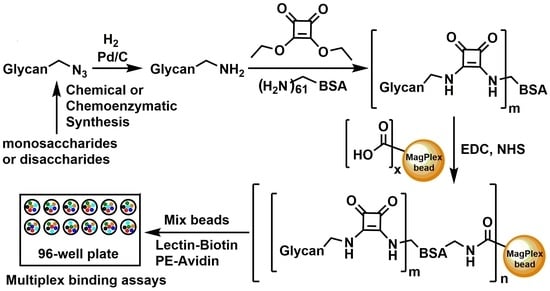
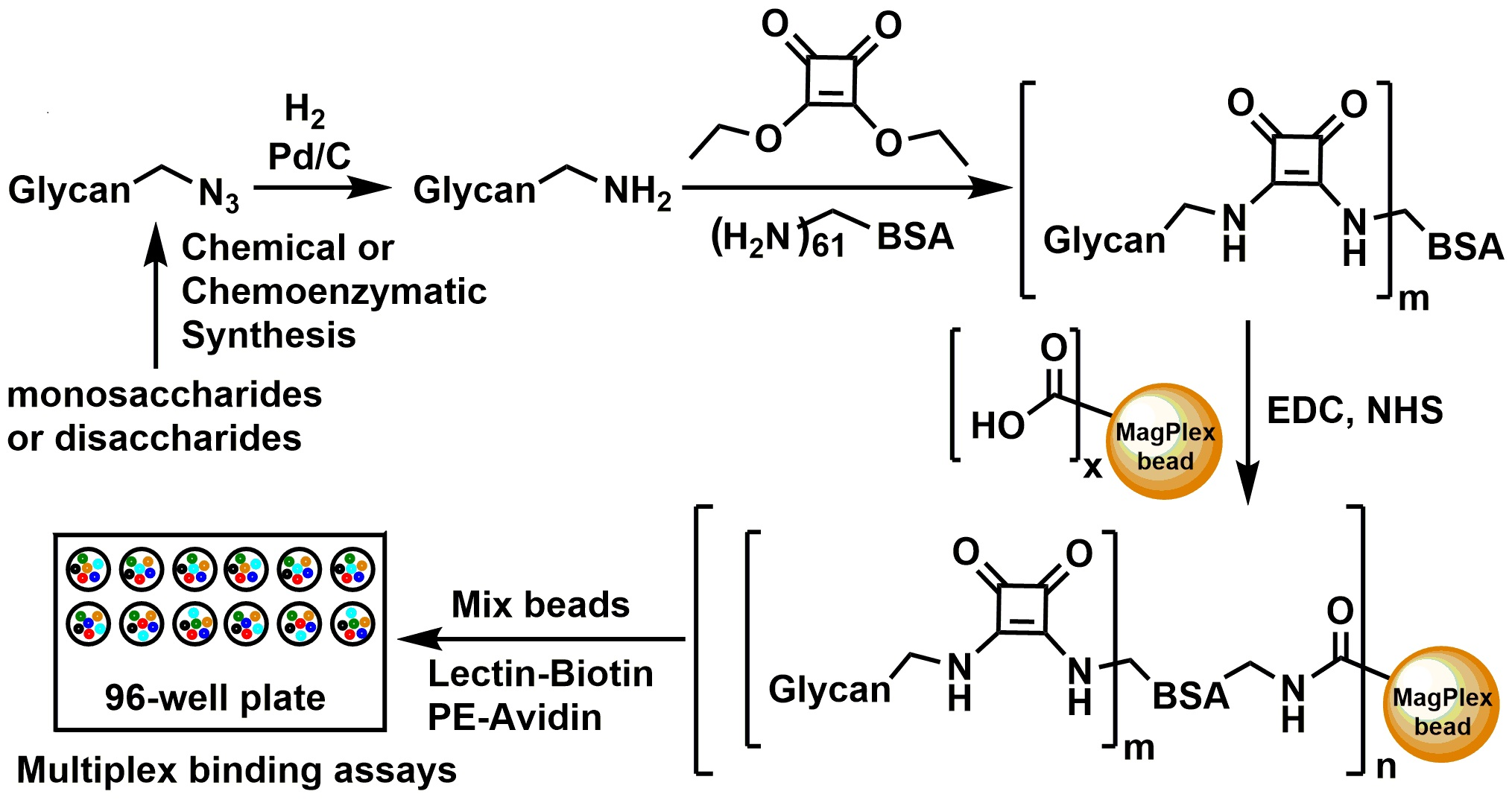
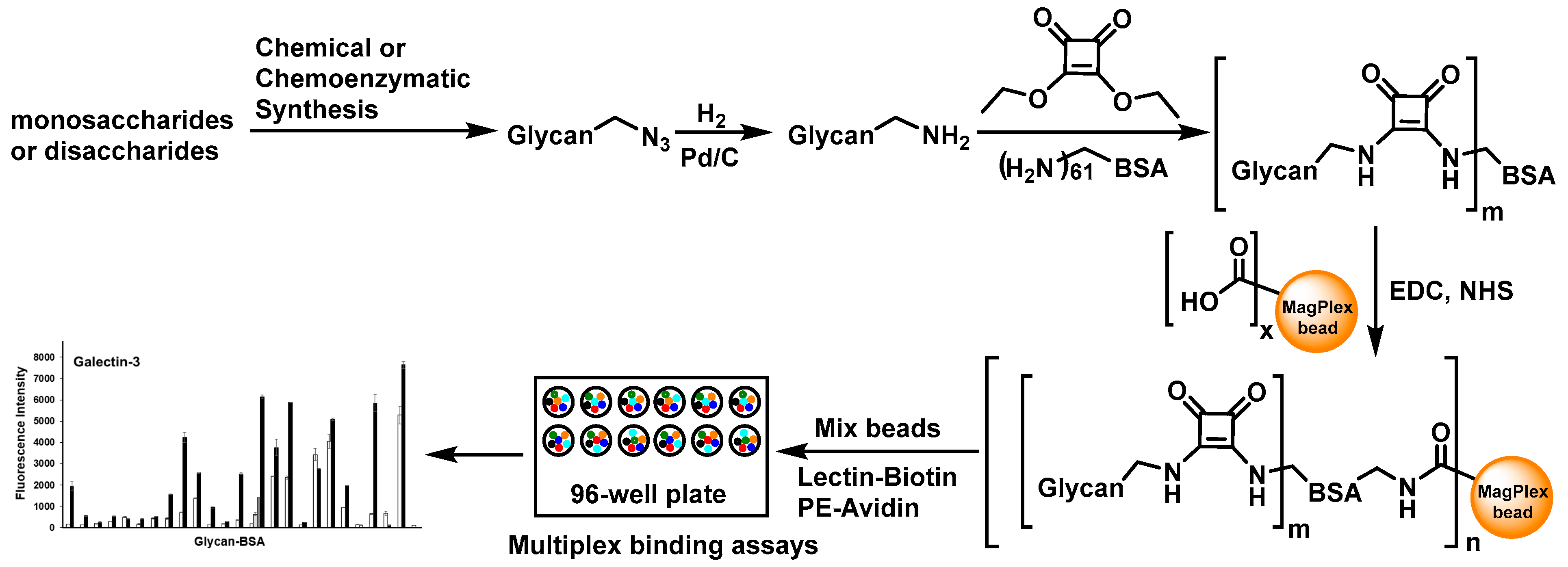
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
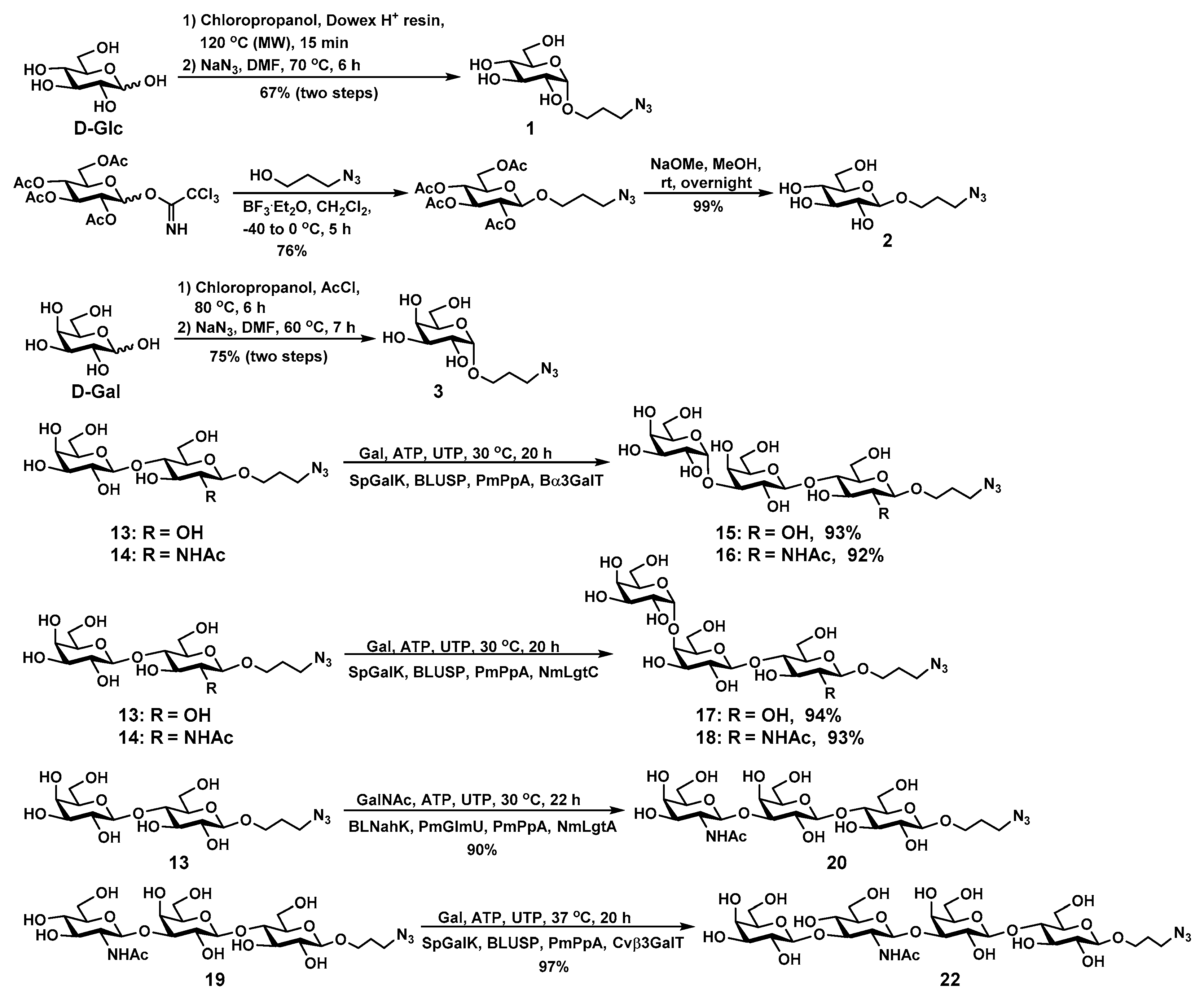
2.1. Glycan Synthesis
2.2. Glycan-BSA Neoglycoprotein Synthesis
2.3. Glycan-BSA Immobilization to MagPlex Beads
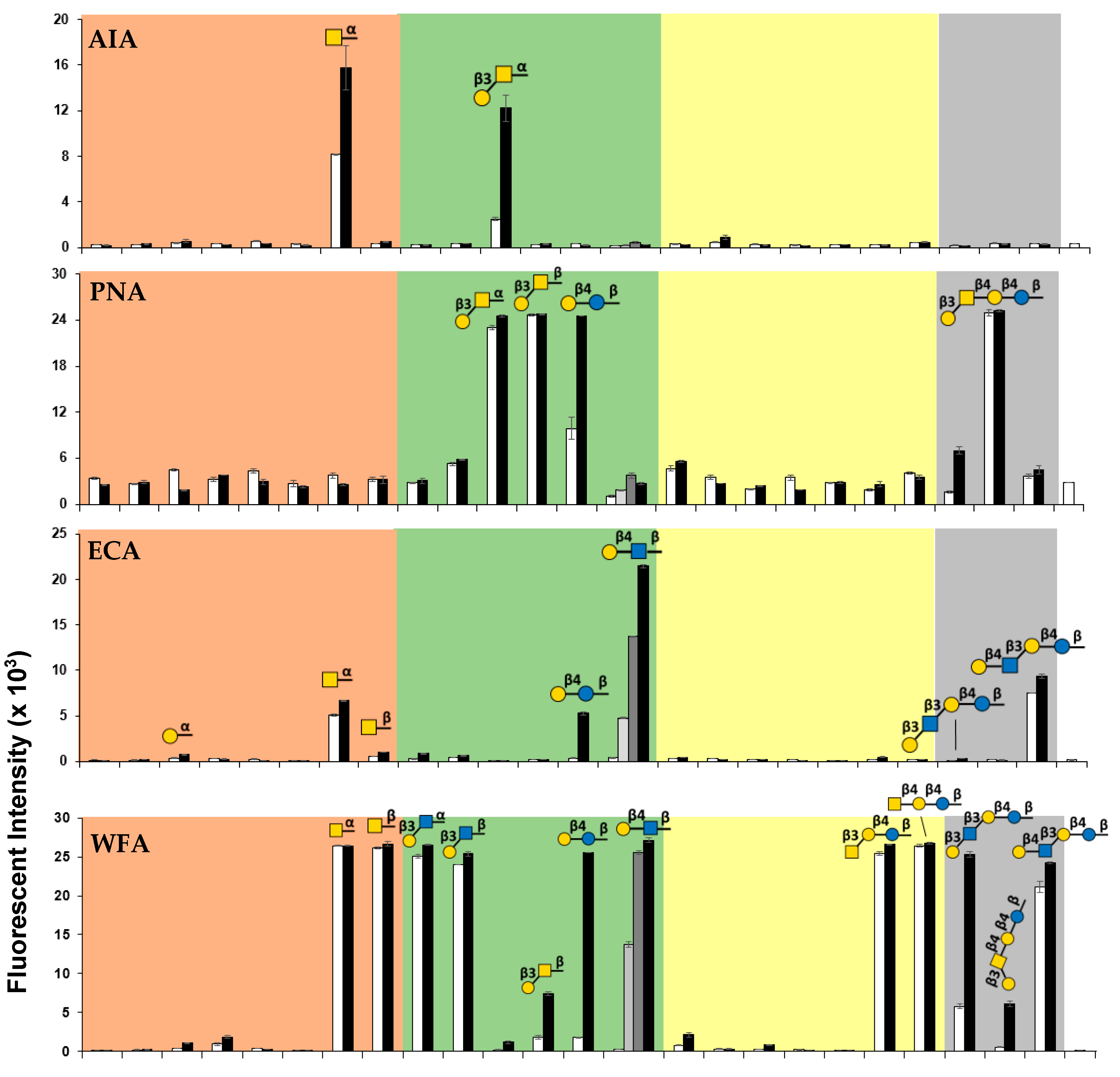
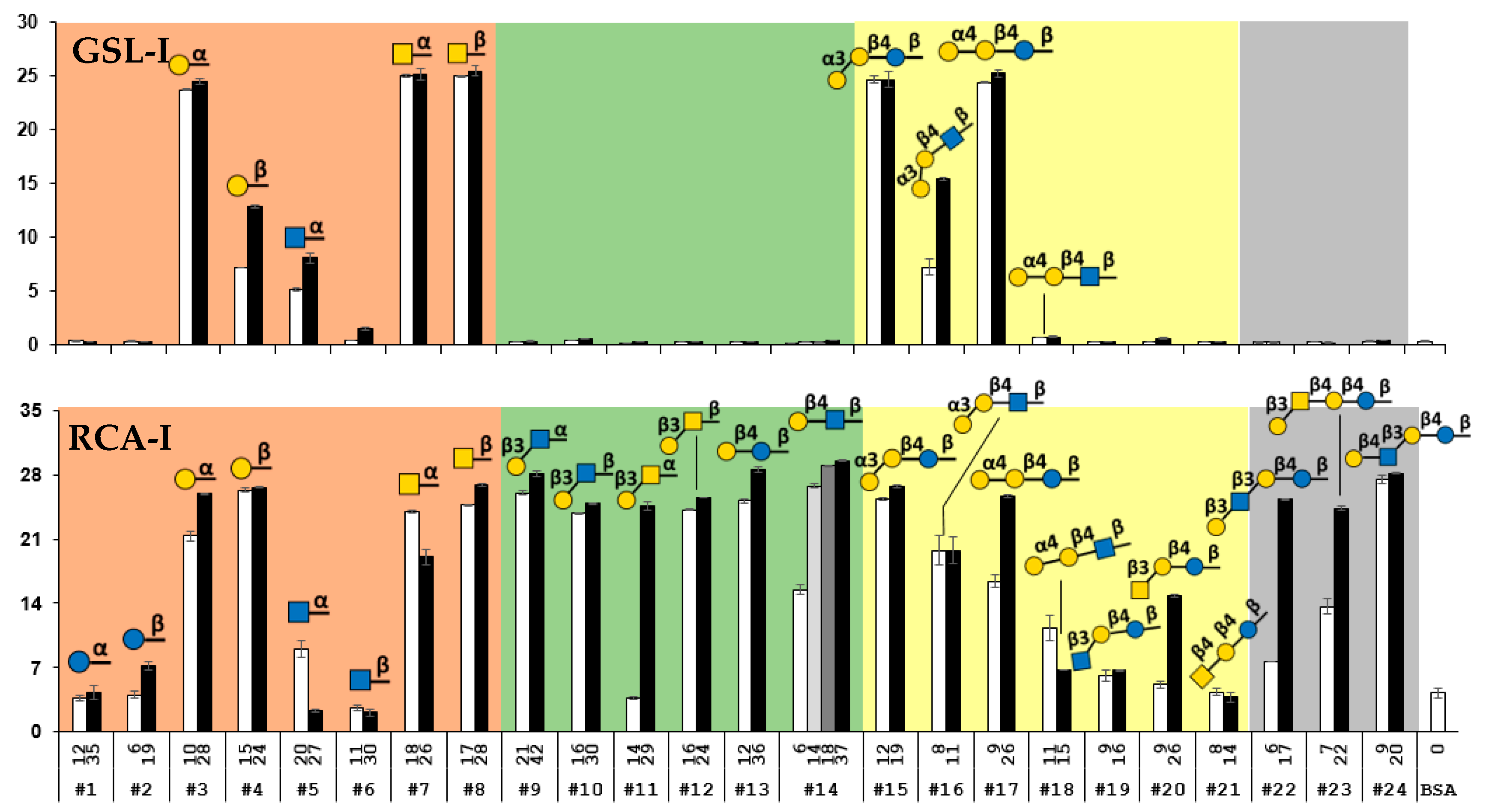
2.4. Suspension Multiplex Array Assays for Plant Lectins
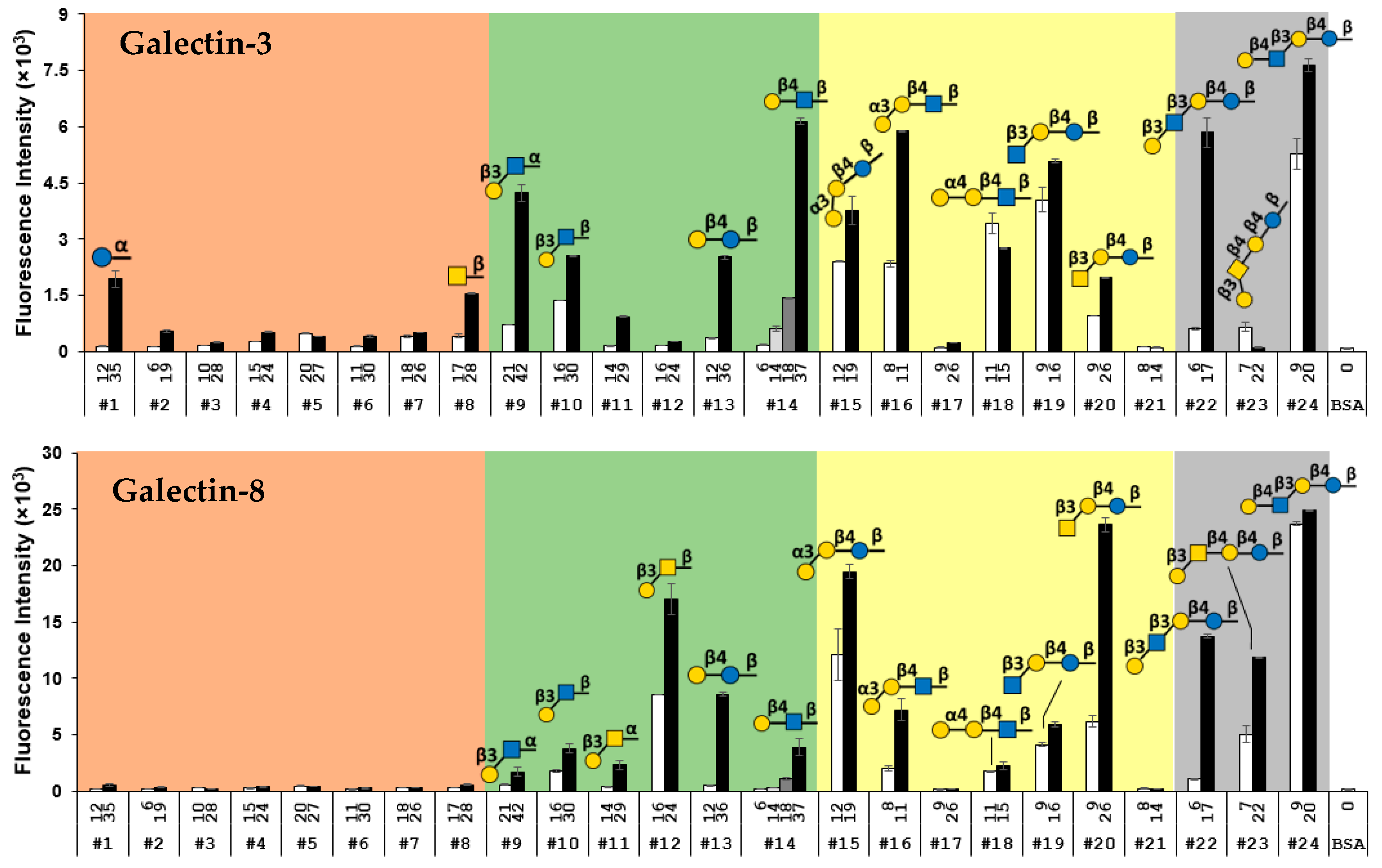
2.5. Multiplex Suspension Array Assays for Recombinant Human Galectin-3 and 8
3. Discussion
4. Materials and Methods
4.1. Materials and Instruments
4.2. Magnetic Fluorescent Beads
4.3. Plant Lectins, Galectins, and Antibodies
4.4. Syntheisis of Glycans
4.5. General Procedures for Converting Propylazide-Tagged Glycans to Glycosyl Propylamines
4.6. General Procedures for Preparing Glycan-Squarate Conjugates
4.7. Coupling Glycan-Squarate to BSA
4.8. Immobilization of Glycan-BSA Neoglycoprotein Conjugates to MagPlex Beads
4.9. Prepare Neoglycoprotein-Immobilized MagPlex Beads Library for Binding Assays
4.10. Multiplex Glycan-Binding Assays for Plant Lectins
4.11. Multiplex Glycan-Binding Assays for Recombinant Human Galectins
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Taylor, M.E.; Drickamer, K.; Schnaar, R.L.; Etzler, M.E.; Varki, A. Discovery and classification of glycan-binding proteins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2015; pp. 361–372. [Google Scholar]
- Van Breedam, W.; Pohlmann, S.; Favoreel, H.W.; de Groot, R.J.; Nauwynck, H.J. Bitter-sweet symphony: Glycan-lectin interactions in virus biology. FEMS Microbiol. Rev. 2014, 38, 598–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imberty, A.; Varrot, A. Microbial recognition of human cell surface glycoconjugates. Curr. Opin. Struct. Biol. 2008, 18, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Marth, J.D.; Grewal, P.K. Mammalian glycosylation in immunity. Nat. Rev. Immunol. 2008, 8, 874–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, J.N.; Saldova, R.; Hamid, U.M.; Rudd, P.M. Evaluation of the serum N-linked glycome for the diagnosis of cancer and chronic inflammation. Proteomics 2008, 8, 3284–3293. [Google Scholar] [CrossRef]
- Cohen, M.; Varki, A. Modulation of glycan recognition by clustered saccharide patches. Int. Rev. Cell Mol. Biol. 2014, 308, 75–125. [Google Scholar] [PubMed]
- Wang, L.; Cummings, R.D.; Smith, D.F.; Huflejt, M.; Campbell, C.T.; Gildersleeve, J.C.; Gerlach, J.Q.; Kilcoyne, M.; Joshi, L.; Serna, S.; et al. Cross-platform comparison of glycan microarray formats. Glycobiology 2014, 24, 507–517. [Google Scholar] [CrossRef] [Green Version]
- Padler-Karavani, V.; Song, X.; Yu, H.; Hurtado-Ziola, N.; Huang, S.; Muthana, S.; Chokhawala, H.A.; Cheng, J.; Verhagen, A.; Langereis, M.A.; et al. Cross-comparison of protein recognition of sialic acid diversity on two novel sialoglycan microarrays. J. Biol. Chem. 2012, 287, 22593–22608. [Google Scholar] [CrossRef] [Green Version]
- Houseman, B.T.; Mrksich, M. Carbohydrate arrays for the evaluation of protein binding and enzymatic modification. Chem. Biol. 2002, 9, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Blixt, O.; Head, S.; Mondala, T.; Scanlan, C.; Huflejt, M.E.; Alvarez, R.; Bryan, M.C.; Fazio, F.; Calarese, D.; Stevens, J.; et al. Printed covalent glycan array for ligand profiling of diverse glycan binding proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 17033–17038. [Google Scholar] [CrossRef] [Green Version]
- Fukui, S.; Feizi, T.; Galustian, C.; Lawson, A.M.; Chai, W. Oligosaccharide microarrays for high-throughput detection and specificity assignments of carbohydrate-protein interactions. Nat. Biotechnol. 2002, 20, 1011–1017. [Google Scholar] [CrossRef]
- Chang, S.H.; Han, J.L.; Tseng, S.Y.; Lee, H.Y.; Lin, C.W.; Lin, Y.C.; Jeng, W.Y.; Wang, A.H.; Wu, C.Y.; Wong, C.H. Glycan array on aluminum oxide-coated glass slides through phosphonate chemistry. J. Am. Chem. Soc. 2010, 132, 13371–13380. [Google Scholar] [CrossRef]
- Ratner, D.M.; Adams, E.W.; Su, J.; O’Keefe, B.R.; Mrksich, M.; Seeberger, P.H. Probing protein-carbohydrate interactions with microarrays of synthetic oligosaccharides. Chembiochem 2004, 5, 379–382. [Google Scholar] [CrossRef]
- Zhang, Y.; Gildersleeve, J.C. General procedure for the synthesis of neoglycoproteins and immobilization on epoxide-modified glass slides. Methods Mol. Biol. 2012, 808, 155–165. [Google Scholar]
- Adams, E.W.; Ueberfeld, J.; Ratner, D.M.; O’Keefe, B.R.; Walt, D.R.; Seeberger, P.H. Encoded fiber-optic microsphere arrays for probing protein-carbohydrate interactions. Angew. Chem. Int. Ed. Engl. 2003, 42, 5317–5320. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ito, S.; Yasukawa, F.; Konami, Y.; Matsumoto, N. Measurement of the carbohydrate-binding specificity of lectins by a multiplexed bead-based flow cytometric assay. Anal. Biochem. 2005, 336, 28–38. [Google Scholar] [CrossRef]
- Purohit, S.; Li, T.; Guan, W.; Song, X.; Song, J.; Tian, Y.; Li, L.; Sharma, A.; Dun, B.; Mysona, D.; et al. Multiplex glycan bead array for high throughput and high content analyses of glycan binding proteins. Nat. Commun. 2018, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettman, J.R.; Davies, T.; Chandler, D.; Oliver, K.G.; Fulton, R.J. Classification and properties of 64 multiplexed microsphere sets. Cytometry 1998, 33, 234–243. [Google Scholar] [CrossRef]
- Elshal, M.F.; McCoy, J.P. Multiplex bead array assays: Performance evaluation and comparison of sensitivity to ELISA. Methods 2006, 38, 317–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, J.P.; Sklar, L.A. Suspension array technology: Evolution of the flat-array paradigm. Trends Biotechnol. 2002, 20, 9–12. [Google Scholar] [CrossRef]
- Lucas, J.L.; Tacheny, E.A.; Ferris, A.; Galusha, M.; Srivastava, A.K.; Ganguly, A.; Williams, P.M.; Sachs, M.C.; Thurin, M.; Tricoli, J.V.; et al. Development and validation of a Luminex assay for detection of a predictive biomarker for PROSTVAC-VF therapy. PLoS ONE 2017, 12, e0182739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temme, J.S.; Campbell, C.T.; Gildersleeve, J.C. Factors contributing to variability of glycan microarray binding profiles. Faraday Discuss. 2019, 219, 90–111. [Google Scholar] [CrossRef] [PubMed]
- Manimala, J.C.; Roach, T.A.; Li, Z.; Gildersleeve, J.C. High-throughput carbohydrate microarray profiling of 27 antibodies demonstrates widespread specificity problems. Glycobiology 2007, 17, 17C–23C. [Google Scholar] [CrossRef]
- Oyelaran, O.; Gildersleeve, J.C. Glycan arrays: Recent advances and future challenges. Curr. Opin. Chem. Biol. 2009, 13, 406–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Chokhawala, H.; Karpel, R.; Yu, H.; Wu, B.; Zhang, J.; Zhang, Y.; Jia, Q.; Chen, X. A multifunctional Pasteurella multocida sialyltransferase: A powerful tool for the synthesis of sialoside libraries. J. Am. Chem. Soc. 2005, 127, 17618–17619. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Huang, S.; Chokhawala, H.; Sun, M.; Zheng, H.; Chen, X. Highly efficient chemoenzymatic synthesis of naturally occurring and non-natural alpha-2,6-linked sialosides: A P. damsela alpha-2,6-sialyltransferase with extremely flexible donor-substrate specificity. Angew. Chem. Int. Ed. Engl. 2006, 45, 3938–3944. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Thon, V.; Lau, K.; Cai, L.; Chen, Y.; Mu, S.; Li, Y.; Wang, P.G.; Chen, X. Highly efficient chemoenzymatic synthesis of beta1-3-linked galactosides. Chem. Commun. 2010, 46, 7507–7509. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zeng, J.; Li, Y.; Thon, V.; Shi, B.; Chen, X. Effective one-pot multienzyme (OPME) synthesis of monotreme milk oligosaccharides and other sialosides containing 4-O-acetyl sialic acid. Org. Biomol. Chem. 2016, 14, 8586–8597. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yu, H.; Yang, X.; Kooner, A.S.; Luu, B.; Chen, X. One-pot multienzyme (OPME) chemoenzymatic synthesis of brain ganglioside glycans with human ST3GAL II expressed in E. coli. Submitted.
- Lau, K.; Thon, V.; Yu, H.; Ding, L.; Chen, Y.; Muthana, M.M.; Wong, D.; Huang, R.; Chen, X. Highly efficient chemoenzymatic synthesis of beta1-4-linked galactosides with promiscuous bacterial beta1-4-galactosyltransferases. Chem. Commun. 2010, 46, 6066–6068. [Google Scholar] [CrossRef] [Green Version]
- Stanley, P.; Cummings, R.D. Structures common to different glycans. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2015; pp. 161–178. [Google Scholar]
- Coombs, P.J.; Taylor, M.E.; Drickamer, K. Two categories of mammalian galactose-binding receptors distinguished by glycan array profiling. Glycobiology 2006, 16, 1C–7C. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Gil, G.C.; Harvey, D.J.; Kim, B.G. Structural analysis of alpha-Gal and new non-Gal carbohydrate epitopes from specific pathogen-free miniature pig kidney. Proteomics 2008, 8, 2596–2610. [Google Scholar] [CrossRef] [PubMed]
- Chen, X. Human milk oligosaccharides (HMOS): Structure, function, and enzyme-catalyzed synthesis. Adv. Carbohydr. Chem. Biochem. 2015, 72, 113–190. [Google Scholar] [PubMed]
- Yu, H.; Li, Y.; Zeng, J.; Thon, V.; Nguyen, D.M.; Ly, T.; Kuang, H.Y.; Ngo, A.; Chen, X. Sequential one-pot multienzyme chemoenzymatic synthesis of glycosphingolipid glycans. J. Org. Chem. 2016, 81, 10809–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Li, J.; Chen, X.; Zhang, Y.; Wang, J.; Guo, Z.; Zhang, W.; Yu, L.; Brew, K.; Wang, P.G. Highly efficient chemoenzymatic synthesis of α-galactosyl epitopes with a recombinant α(1,3)-galactosyltransferase. J. Am. Chem. Soc. 1998, 120, 6635–6638. [Google Scholar] [CrossRef]
- Chen, M.; Chen, L.L.; Zou, Y.; Xue, M.; Liang, M.; Jin, L.; Guan, W.Y.; Shen, J.; Wang, W.; Wang, L.; et al. Wide sugar substrate specificity of galactokinase from Streptococcus pneumoniae TIGR4. Carbohydr. Res. 2011, 346, 2421–2425. [Google Scholar] [CrossRef] [PubMed]
- Muthana, M.M.; Qu, J.; Li, Y.; Zhang, L.; Yu, H.; Ding, L.; Malekan, H.; Chen, X. Efficient one-pot multienzyme synthesis of UDP-sugars using a promiscuous UDP-sugar pyrophosphorylase from Bifidobacterium longum (BLUSP). Chem. Commun. 2012, 48, 2728–2730. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, J.; Kowal, P.; Liu, Z.; Andreana, P.R.; Lu, Y.; Wang, P.G. Transferring a biosynthetic cycle into a productive Escherichia coli strain: Large-scale synthesis of galactosides. J. Am. Chem. Soc. 2001, 123, 8866–8867. [Google Scholar] [CrossRef]
- Galili, U. The alpha-gal epitope and the anti-Gal antibody in xenotransplantation and in cancer immunotherapy. Immunol. Cell Biol. 2005, 83, 674–686. [Google Scholar] [CrossRef]
- Galili, U.; Clark, M.R.; Shohet, S.B.; Buehler, J.; Macher, B.A. Evolutionary relationship between the natural anti-Gal antibody and the Gal alpha 1-3Gal epitope in primates. Proc. Natl. Acad. Sci. USA 1987, 84, 1369–1373. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Mattner, J.; Cantu, C., 3rd; Schrantz, N.; Yin, N.; Gao, Y.; Sagiv, Y.; Hudspeth, K.; Wu, Y.P.; Yamashita, T.; et al. Lysosomal glycosphingolipid recognition by NKT cells. Science 2004, 306, 1786–1789. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, G.D.; Fodor, E.; Vanmaele, R. Investigation of Shiga-like toxin binding to chemically synthesized oligosaccharide sequences. J. Infect. Dis. 1991, 164, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Bruce, L.J. Molecular mechanism of P1 antigen expression. Blood 2018, 131, 1505–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolaou, K.C.; Caulfield, T.J.; Katoaka, H. Total synthesis of globotriaosylceramide (Gb3) and lysoglobotriaosylceramide (lysoGb3). Carbohydr. Res. 1990, 202, 177–191. [Google Scholar] [CrossRef]
- Blixt, O.; van Die, I.; Norberg, T.; van den Eijnden, D.H. High-level expression of the Neisseria meningitidis lgtA gene in Escherichia coli and characterization of the encoded N-acetylglucosaminyltransferase as a useful catalyst in the synthesis of GlcNAc beta 1→3Gal and GalNAc beta 1→3Gal linkages. Glycobiology 1999, 9, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yu, H.; Chen, Y.; Lau, K.; Cai, L.; Cao, H.; Tiwari, V.K.; Qu, J.; Thon, V.; Wang, P.G.; et al. Substrate promiscuity of N-acetylhexosamine 1-kinases. Molecules 2011, 16, 6396–6407. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Thon, V.; Li, Y.; Yu, H.; Ding, L.; Lau, K.; Qu, J.; Hie, L.; Chen, X. One-pot three-enzyme synthesis of UDP-GlcNAc derivatives. Chem. Commun. 2011, 47, 10815–10817. [Google Scholar] [CrossRef]
- Tseng, H.K.; Su, Y.Y.; Chang, T.W.; Liu, H.C.; Li, P.J.; Chiang, P.Y.; Lin, C.C. Acceptor-mediated regioselective enzyme catalyzed sialylation: Chemoenzymatic synthesis of GAA-7 ganglioside glycan. Chem. Commun. 2021, 57, 3468–3471. [Google Scholar] [CrossRef] [PubMed]
- Tamai, H.; Imamura, A.; Ogawa, J.; Ando, H.; Ishida, H.; Kiso, M. First total synthesis of ganglioside GAA-7 from starfish Asterias amurensis versicolor. Eur. J. Org. Chem. 2015, 2015, 5199–5211. [Google Scholar] [CrossRef]
- McArthur, J.B.; Yu, H.; Chen, X. A bacterial β1-3-galactosyltransferase enables multigram-scale synthesis of human milk lacto-N-tetraose (LNT) and its fucosides. ACS Catal. 2019, 9, 10721–10726. [Google Scholar] [CrossRef]
- Bode, L. Human milk oligosaccharides: Every baby needs a sugar mama. Glycobiology 2012, 22, 1147–1162. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Chokhawala, H.A.; Varki, A.; Chen, X. Efficient chemoenzymatic synthesis of biotinylated human serum albumin-sialoglycoside conjugates containing O-acetylated sialic acids. Org. Biomol. Chem. 2007, 5, 2458–2463. [Google Scholar] [CrossRef] [PubMed]
- Tietze, L.F.; Schroter, C.; Gabius, S.; Brinck, U.; Goerlach-Graw, A.; Gabius, H.J. Conjugation of p-aminophenyl glycosides with squaric acid diester to a carrier protein and the use of neoglycoprotein in the histochemical detection of lectins. Bioconjug. Chem. 1991, 2, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Blixt, O.; Norberg, T. Enzymatic glycosylation of reducing oligosaccharides linked to a solid phase or a lipid via a cleavable squarate linker. Carbohydr. Res. 1999, 319, 80–91. [Google Scholar] [CrossRef]
- Kamath, V.P.; Diedrich, P.; Hindsgaul, O. Use of diethyl squarate for the coupling of oligosaccharide amines to carrier proteins and characterization of the resulting neoglycoproteins by MALDI-TOF mass spectrometry. Glycoconj. J. 1996, 13, 315–319. [Google Scholar] [CrossRef]
- Nitz, M.; Bundle, D.R. Synthesis of di- to hexasaccharide 1,2-linked beta-mannopyranan oligomers, a terminal S-linked tetrasaccharide congener and the corresponding BSA glycoconjugates. J. Org. Chem. 2001, 66, 8411–8423. [Google Scholar] [CrossRef]
- Wurm, F.R.; Klok, H.-A. Be squared: Expanding the horizon of squaric acid-mediated conjugations. Chem. Soc. Rev. 2013, 42, 8220–8236. [Google Scholar] [CrossRef]
- Cummings, R.D.; Schnaar, R.L. R-type lectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2015; pp. 401–412. [Google Scholar]
- Green, E.D.; Brodbeck, R.M.; Baenziger, J.U. Lectin affinity high-performance liquid chromatography. Interactions of N-glycanase-released oligosaccharides with Ricinus communis agglutinin I and Ricinus communis agglutinin II. J. Biol. Chem. 1987, 262, 12030–12039. [Google Scholar] [CrossRef]
- Li, N.; Chow, A.M.; Ganesh, H.V.; Brown, I.R.; Kerman, K. Quantum dot based fluorometric detection of cancer TF-antigen. Anal. Chem. 2013, 85, 9699–9704. [Google Scholar] [CrossRef]
- Duk, M.; Lisowska, E.; Wu, J.H.; Wu, A.M. The biotin/avidin-mediated microtiter plate lectin assay with the use of chemically modified glycoprotein ligand. Anal. Biochem. 1994, 221, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Vijayan, M.; Surolia, A. Imparting exquisite specificity to peanut agglutinin for the tumor-associated Thomsen-Friedenreich antigen by redesign of its combining site. J. Biol. Chem. 1996, 271, 21209–21213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iglesias, J.L.; Lis, H.; Sharon, N. Purification and properties of a D-galactose/N-acetyl-D-galactosamine-specific lectin from Erythrina cristagalli. Eur. J. Biochem. 1982, 123, 247–252. [Google Scholar] [CrossRef]
- Kurokawa, T.; Tsuda, M.; Sugino, Y. Purification and characterization of a lectin from Wistaria floribunda seeds. J. Biol. Chem. 1976, 251, 5686–5693. [Google Scholar] [CrossRef]
- Qiu, Y.; Tian, S.; Gu, L.; Hildreth, M.; Zhou, R. Identification of surface polysaccharides in akinetes, heterocysts and vegetative cells of Anabaena cylindrica using fluorescein-labeled lectins. Arch. Microbiol. 2019, 201, 17–25. [Google Scholar] [CrossRef]
- Sato, T.; Tateno, H.; Kaji, H.; Chiba, Y.; Kubota, T.; Hirabayashi, J.; Narimatsu, H. Engineering of recombinant Wisteria floribunda agglutinin specifically binding to GalNAcβ1,4GlcNAc (LacdiNAc). Glycobiology 2017, 27, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Hartig, W.; Brauer, K.; Bruckner, G. Wisteria floribunda agglutinin-labelled nets surround parvalbumin-containing neurons. Neuroreport 1992, 3, 869–872. [Google Scholar] [CrossRef]
- Matsuda, A.; Kuno, A.; Kawamoto, T.; Matsuzaki, H.; Irimura, T.; Ikehara, Y.; Zen, Y.; Nakanuma, Y.; Yamamoto, M.; Ohkohchi, N.; et al. Wisteria floribunda agglutinin-positive mucin 1 is a sensitive biliary marker for human cholangiocarcinoma. Hepatology 2010, 52, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, H.; Sato, T. Wisteria floribunda agglutinin positive glycobiomarkers: A unique lectin as a serum biomarker probe in various diseases. Expert Rev. Proteom. 2018, 15, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Schick, B.; Habermann, F.; Sinowatz, F. Histochemical detection of glycoconjugates in the canine epididymis. Anat. Histol. Embryol. 2009, 38, 122–127. [Google Scholar] [CrossRef]
- Yu, Y.; Mishra, S.; Song, X.; Lasanajak, Y.; Bradley, K.C.; Tappert, M.M.; Air, G.M.; Steinhauer, D.A.; Halder, S.; Cotmore, S.; et al. Functional glycomic analysis of human milk glycans reveals the presence of virus receptors and embryonic stem cell biomarkers. J. Biol. Chem. 2012, 287, 44784–44799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barondes, S.H.; Castronovo, V.; Cooper, D.N.; Cummings, R.D.; Drickamer, K.; Feizi, T.; Gitt, M.A.; Hirabayashi, J.; Hughes, C.; Kasai, K.; et al. Galectins: A family of animal beta-galactoside-binding lectins. Cell 1994, 76, 597–598. [Google Scholar] [CrossRef]
- Cummings, R.D.; Liu, F.T.; Vasta, G.R. Galectins. In Essentials of Glycobiology; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Darvill, A.G., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor: New York, NY, USA, 2015; pp. 469–480. [Google Scholar]
- Hirabayashi, J.; Hashidate, T.; Arata, Y.; Nishi, N.; Nakamura, T.; Hirashima, M.; Urashima, T.; Oka, T.; Futai, M.; Muller, W.E.; et al. Oligosaccharide specificity of galectins: A search by frontal affinity chromatography. Biochim. Biophys. Acta 2002, 1572, 232–254. [Google Scholar] [CrossRef]
- Fred Brewer, C. Binding and cross-linking properties of galectins. Biochim. Biophys. Acta 2002, 1572, 255–262. [Google Scholar] [CrossRef]
- Dam, T.K.; Gerken, T.A.; Brewer, C.F. Thermodynamics of multivalent carbohydrate-lectin cross-linking interactions: Importance of entropy in the bind and jump mechanism. Biochemistry 2009, 48, 3822–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. Engl. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Godula, K. Following sugar patterns in search of galectin function. Proc. Natl. Acad. Sci. USA 2018, 115, 2548–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Ludwig, A.K.; Romano, C.; Buzzacchera, I.; Sherman, S.E.; Vetro, M.; Vertesy, S.; Kaltner, H.; Reed, E.H.; Moller, M.; et al. Exploring functional pairing between surface glycoconjugates and human galectins using programmable glycodendrimersomes. Proc. Natl. Acad. Sci. USA 2018, 115, E2509–E2518. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Zheng, Y.; Xu, D.; Sun, Z.; Yang, H.; Yin, Q. Galectin-3: A key player in microglia-mediated neuroinflammation and Alzheimer’s disease. Cell Biosci. 2021, 11, 78. [Google Scholar] [CrossRef] [PubMed]
- Blanda, V.; Bracale, U.M.; Di Taranto, M.D.; Fortunato, G. Galectin-3 in cardiovascular diseases. Int. J. Mol. Sci. 2020, 21, 9232. [Google Scholar] [CrossRef]
- Sciacchitano, S.; Lavra, L.; Morgante, A.; Ulivieri, A.; Magi, F.; De Francesco, G.P.; Bellotti, C.; Salehi, L.B.; Ricci, A. Galectin-3: One molecule for an alphabet of diseases, from A to Z. Int. J. Mol. Sci. 2018, 19, 379. [Google Scholar] [CrossRef] [Green Version]
- Tribulatti, M.V.; Carabelli, J.; Prato, C.A.; Campetella, O. Galectin-8 in the onset of the immune response and inflammation. Glycobiology 2020, 30, 134–142. [Google Scholar] [CrossRef]
- Horlacher, T.; Oberli, M.A.; Werz, D.B.; Krock, L.; Bufali, S.; Mishra, R.; Sobek, J.; Simons, K.; Hirashima, M.; Niki, T.; et al. Determination of carbohydrate-binding preferences of human galectins with carbohydrate microarrays. Chembiochem 2010, 11, 1563–1573. [Google Scholar] [CrossRef]
- Song, X.; Xia, B.; Stowell, S.R.; Lasanajak, Y.; Smith, D.F.; Cummings, R.D. Novel fluorescent glycan microarray strategy reveals ligands for galectins. Chem. Biol. 2009, 16, 36–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowell, S.R.; Arthur, C.M.; Mehta, P.; Slanina, K.A.; Blixt, O.; Leffler, H.; Smith, D.F.; Cummings, R.D. Galectin-1, -2, and -3 exhibit differential recognition of sialylated glycans and blood group antigens. J. Biol. Chem. 2008, 283, 10109–10123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Yasukawa, F.; Ito, S. Measurement of the sugar-binding specificity of lectins using multiplexed bead-based suspension arrays. Methods Mol. Biol. 2007, 381, 401–409. [Google Scholar] [PubMed]
- Yamamoto, K. Carbohydrate-binding specificity of lectins using multiplexed glyco-bead array. Methods Mol. Biol. 2014, 1200, 319–326. [Google Scholar] [PubMed]
- Iihara, H.; Niwa, T.; Shah, M.M.; Nhung, P.H.; Song, S.X.; Hayashi, M.; Ohkusa, K.; Itoh, Y.; Makino, S.; Ezaki, T. Rapid multiplex immunofluorescent assay to detect antibodies against Burkholderia pseudomallei and taxonomically closely related nonfermenters. Jpn. J. Infect. Dis. 2007, 60, 230–234. [Google Scholar]
- Berger, S.S.; Lauritsen, K.T.; Boas, U.; Lind, P.; Andresen, L.O. Simultaneous detection of antibodies to five Actinobacillus pleuropneumoniae serovars using bead-based multiplex analysis. J. Vet. Diagn. Investig. 2017, 29, 797–804. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, R.W.; Clarkson, K.; Kordis, A.A.; Oaks, E.V. Multiplexed immunoassay to assess Shigella-specific antibody responses. J. Immunol. Methods 2013, 393, 18–29. [Google Scholar] [CrossRef]
- Gray, B.M. ELISA methodology for polysaccharide antigens: Protein coupling of polysaccharides for adsorption to plastic tubes. J. Immunol. Methods 1979, 28, 187–192. [Google Scholar] [CrossRef]
- Pickering, J.W.; Martins, T.B.; Greer, R.W.; Schroder, M.C.; Astill, M.E.; Litwin, C.M.; Hildreth, S.W.; Hill, H.R. A multiplexed fluorescent microsphere immunoassay for antibodies to pneumococcal capsular polysaccharides. Am. J. Clin. Pathol. 2002, 117, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Pickering, J.W.; Hill, H.R. Measurement of antibodies to pneumococcal polysaccharides with Luminex xMAP microsphere-based liquid arrays. In Carbohydrate Microarrays: Methods and Protocols; Chevolot, Y., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 361–375. [Google Scholar]
- Bujacz, A.; Zielinski, K.; Sekula, B. Structural studies of bovine, equine, and leporine serum albumin complexes with naproxen. Proteins 2014, 82, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Bundle, D.R.; Tam, P.H.; Tran, H.A.; Paszkiewicz, E.; Cartmell, J.; Sadowska, J.M.; Sarkar, S.; Joe, M.; Kitov, P.I. Oligosaccharides and peptide displayed on an amphiphilic polymer enable solid phase assay of hapten specific antibodies. Bioconjug. Chem. 2014, 25, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Bocker, S.; Laaf, D.; Elling, L. Galectin binding to neo-glycoproteins: LacDiNAc conjugated BSA as ligand for human galectin-3. Biomolecules 2015, 5, 1671–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.F.; Song, X.; Cummings, R.D. Use of glycan microarrays to explore specificity of glycan-binding proteins. Methods Enzymol. 2010, 480, 417–444. [Google Scholar]
- Bornaghi, L.F.; Poulsen, S.-A. Microwave-accelerated Fischer glycosylation. Tetrahedron Lett. 2005, 46, 3485–3488. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Cao, X.; Xian, M.; Fang, L.; Cai, T.B.; Ji, J.J.; Tunac, J.B.; Sun, D.; Wang, P.G. Synthesis and enzyme-specific activation of carbohydrate−geldanamycin conjugates with potent anticancer activity. J. Med. Chem. 2005, 48, 645–652. [Google Scholar] [CrossRef]
- Tietze, L.F.; Arlt, M.; Beller, M.; Gl üsenkamp, K.-H.; Jähde, E.; Rajewsky, M.F. Anticancer agents, 15. Squaric acid diethyl ester: A new coupling reagent for the formation of drug biopolymer conjugates. Synthesis of squaric acid ester amides and diamides. Chem. Ber. 1991, 124, 1215–1221. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Yu, H.; Bai, Y.; Mishra, B.; Yang, X.; Wang, J.; Yu, E.B.; Li, R.; Chen, X. A Neoglycoprotein-Immobilized Fluorescent Magnetic Bead Suspension Multiplex Array for Galectin-Binding Studies. Molecules 2021, 26, 6194. https://doi.org/10.3390/molecules26206194
Zhang L, Yu H, Bai Y, Mishra B, Yang X, Wang J, Yu EB, Li R, Chen X. A Neoglycoprotein-Immobilized Fluorescent Magnetic Bead Suspension Multiplex Array for Galectin-Binding Studies. Molecules. 2021; 26(20):6194. https://doi.org/10.3390/molecules26206194
Chicago/Turabian StyleZhang, Libo, Hai Yu, Yuanyuan Bai, Bijoyananda Mishra, Xiaoxiao Yang, Jing Wang, Evan B. Yu, Riyao Li, and Xi Chen. 2021. "A Neoglycoprotein-Immobilized Fluorescent Magnetic Bead Suspension Multiplex Array for Galectin-Binding Studies" Molecules 26, no. 20: 6194. https://doi.org/10.3390/molecules26206194
APA StyleZhang, L., Yu, H., Bai, Y., Mishra, B., Yang, X., Wang, J., Yu, E. B., Li, R., & Chen, X. (2021). A Neoglycoprotein-Immobilized Fluorescent Magnetic Bead Suspension Multiplex Array for Galectin-Binding Studies. Molecules, 26(20), 6194. https://doi.org/10.3390/molecules26206194