
Clarification on the Reactivity of Diaryl Diselenides toward Hexacyclohexyldilead under Light


Abstract
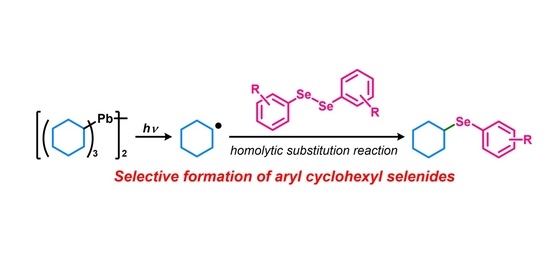
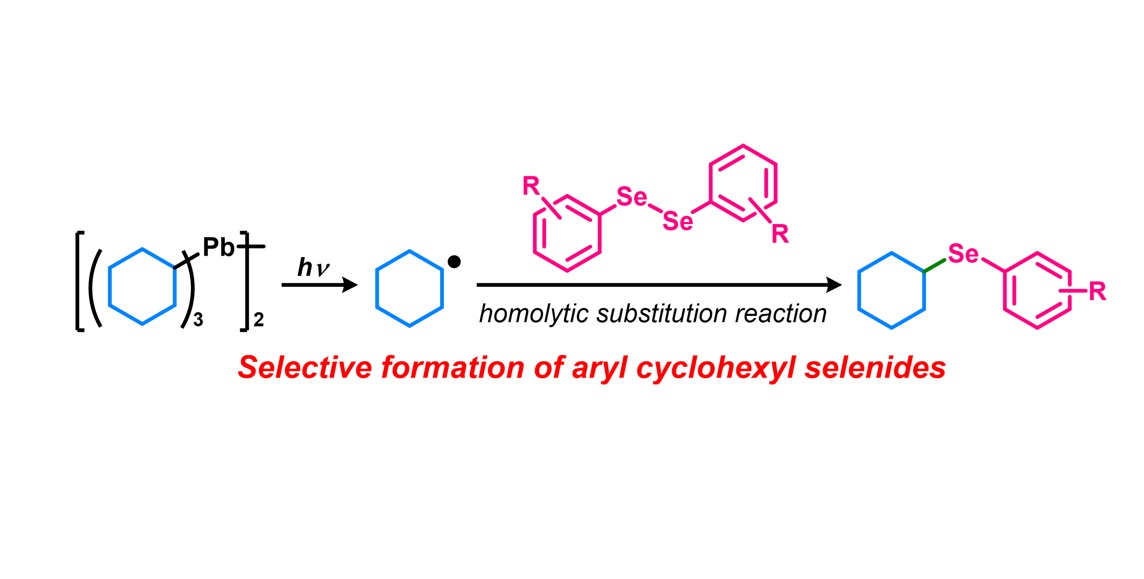
:
1. Introduction
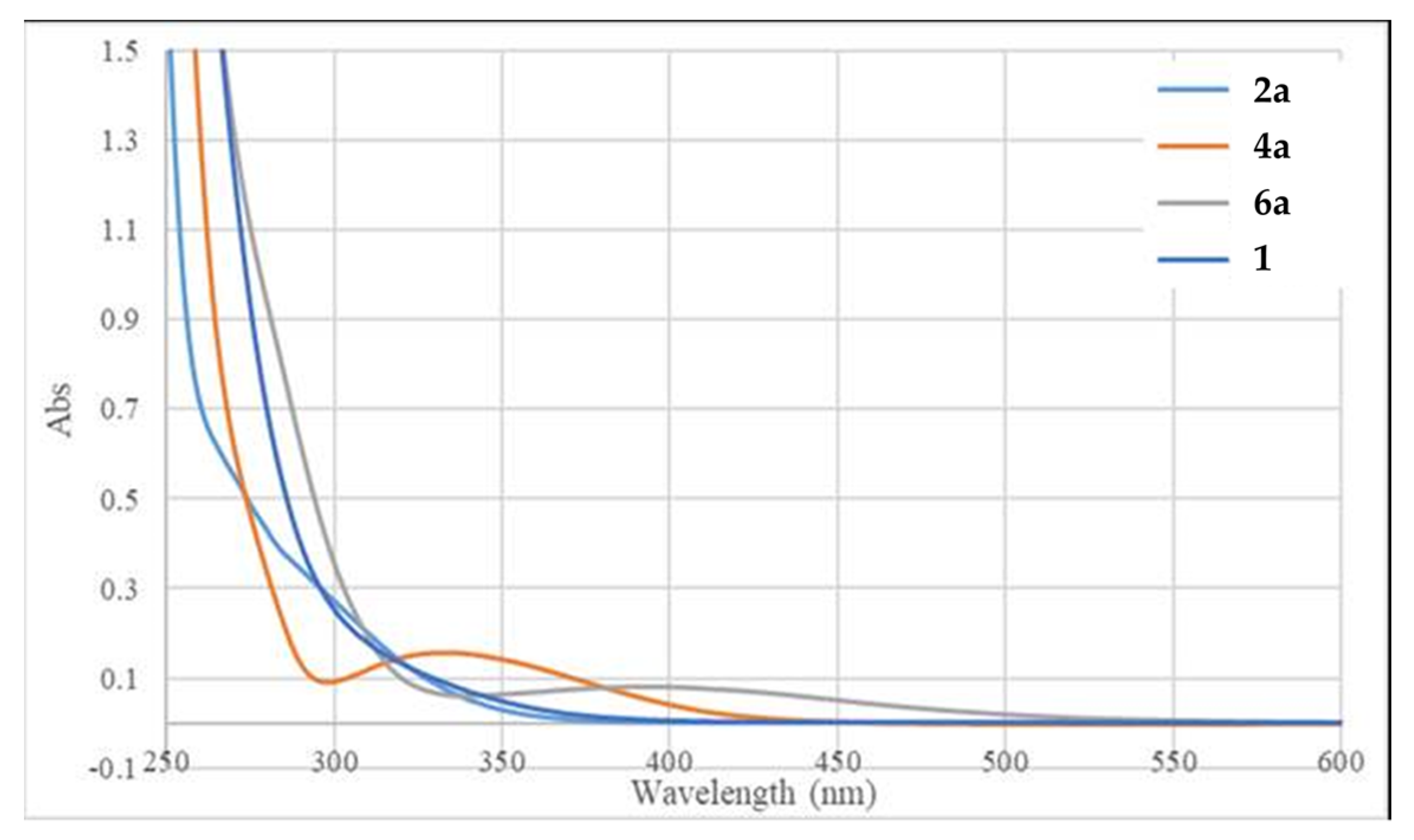
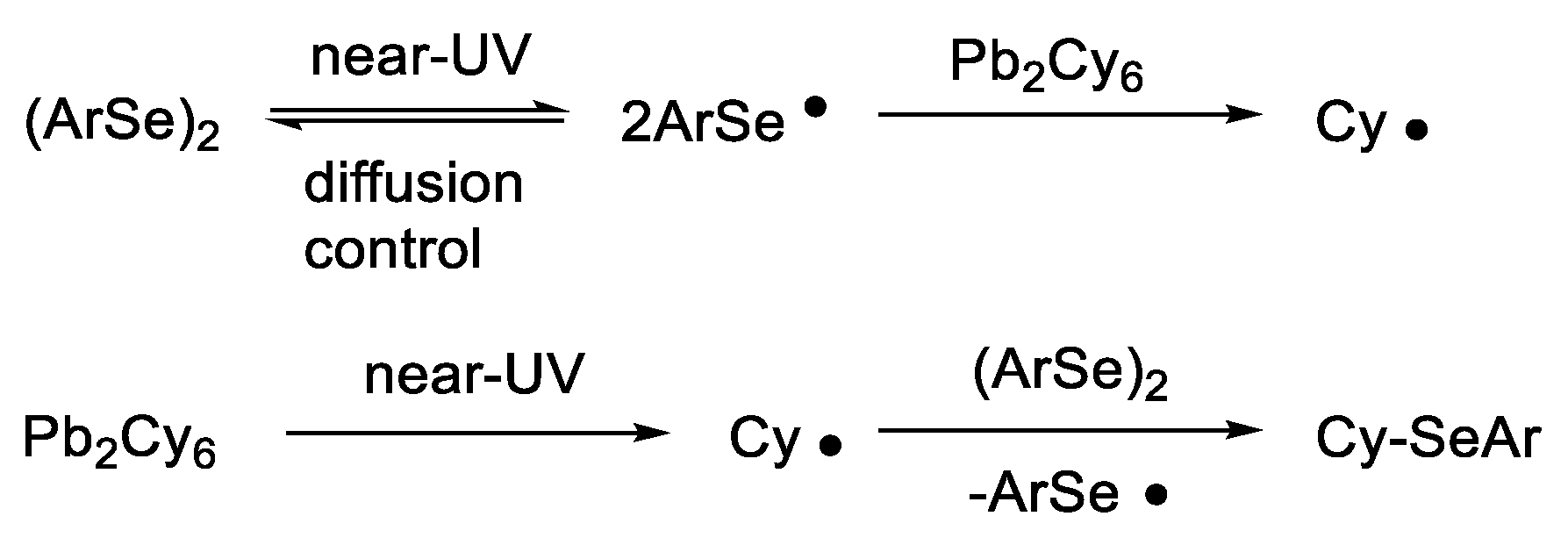
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Procedure for Synthesis of Hexacyclohexyldilead 1
3.3. General Procedure for Synthesis of Diaryl Diselenides
3.4. General Procedure for the Reaction of Diaryl Diselenides with Hexacyclohexyldilead
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Coronado, E.; Forment-Aliaga, A.; Galán-Mascarós, J.R.; Giménez-Saiz, C.; Gómez-García, C.J.; Martinéz-Ferrero, E.; Nuez, A.; Romero, F.M. Multifunctional molecular materials. Solid State Sci. 2003, 5, 917–924. [Google Scholar] [CrossRef]
- Palomba, M.; Franco Coelho Dias, I.; Rosati, O.; Marini, F. Modern Synthetic Strategies with Organoselenium Reagents: A Focus on Vinyl Selenones. Molecules 2021, 26, 3148. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.; Moberg, C. Element–Element Addition to Alkynes Catalyzed by the Group 10 Metals. Chem. Rev. 1999, 99, 3435–3461. [Google Scholar] [CrossRef] [PubMed]
- Han, L.-B.; Tanaka, M. Transition metal-catalysed addition reactions of H-heteroatom and inter-heteroatom bonds to carbon–carbon unsaturated linkages via oxidative additions. Chem. Commun. 1999, 395–402. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-Metal-Catalyzed Addition of Heteroatom–Hydrogen Bonds to Alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef] [PubMed]
- Beletskaya, I.; Moberg, C. Element–Element Additions to Unsaturated Carbon–Carbon Bonds Catalyzed by Transition Metal Complexes. Chem. Rev. 2006, 106, 2320–2354. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C–S, C–Se, and C–Te Bond Formation via Cross-Coupling and Atom-Economic Addition Reactions. Chem. Rev. 2011, 111, 1596–1636. [Google Scholar] [CrossRef]
- Shen, C.; Zhang, P.; Sun, Q.; Bai, S.; Hor, T.S.A.; Liu, X. Recent advances in C–S bond formation via C–H bond functionalization and decarboxylation. Chem. Soc. Rev. 2015, 44, 291–314. [Google Scholar] [CrossRef] [Green Version]
- Ansell, M.B.; Navarro, O.; Spencer, J. Transition metal catalyzed element-element’ additions to alkynes. Coord. Chem. Rev. 2017, 336, 54–77. [Google Scholar] [CrossRef]
- Kawaguchi, S.-i.; Yamamoto, Y.; Ogawa, A. Catalytic synthesis of sulfur and phosphorus compounds via atom-economic reactions. Mendeleev Commun. 2020, 30, 129–138. [Google Scholar] [CrossRef]
- Chiummiento, L.; D’Orsi, R.; Funicello, M.; Lupattelli, P. Last Decade of Unconventional Methodologies for the Synthesis of Substituted Benzofurans. Molecules 2020, 25, 2327. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, P.S.; Peglow, T.J.; Penteado, F.; Bagnoli, L.; Perin, G.; Lenardão, E.J. Recent Advances in the Synthesis of Selenophenes and Their Derivatives. Molecules 2020, 25, 5907. [Google Scholar] [CrossRef] [PubMed]
- Li, G.L.; Huo, X.H.; Jiang, X.Y.; Zhang, W.B. Asymmetric synthesis of allylic compounds via hydrofunctionalisation and difunctionalisation of dienes, allenes, and alkynes. Chem. Soc. Rev. 2020, 49, 2060–2118. [Google Scholar] [CrossRef]
- Sundaravelu, N.; Sangeetha, S.; Sekar, G. Metal-catalyzed C–S bond formation using sulfur surrogates. Org. Biomol. Chem. 2021, 19, 1459–1482. [Google Scholar] [CrossRef]
- Miyaura, N. Hydroboration, Diboration, Silylboration, and Stannylboration. In Catalytic Heterofunctionalization; Togni, A., Grutzmacher, H., Eds.; Wiley-VCH: Weinheim, Germany, 2001; pp. 1–32. [Google Scholar]
- Brunet, J.J.; Neibecker, D. Catalytic Hydroamination of Unsaturated Carbon− Carbon Bonds. In Catalytic Heterofunctionalization; Togni, A., Grutzmacher, H., Eds.; Wiley-VCH: Weinheim, Germany, 2001; pp. 91–141. [Google Scholar]
- Ogawa, A. Transition-Metal-Catalyzed S–H and Se–H to Unsaturated Molecules. Top. Organomet. Chem. 2013, 43, 325–360. [Google Scholar]
- Nomoto, A.; Ogawa, A. Preparative Uses of Organoselenium and Organotellurium Compounds. In The Chemistry of Organic Selenium and Tellurium Compounds; Rappoport, Z., Ed.; Wiley: Chichester, UK, 2012; Volume 3, pp. 623–688. [Google Scholar]
- Ogawa, A.; Tamai, T.; Mitamura, T.; Nomoto, A. Highly selective introduction of heteroatom groups to isocyanides and its application to electrocyclic reactions. Pure Appl. Chem. 2013, 85, 785–799. [Google Scholar] [CrossRef]
- Nomoto, A.; Higuchi, Y.; Kobiki, Y.; Ogawa, A. Synthesis of selenium compounds by free radical addition based on visible-light-activated Se–Se bond cleavage. Mini Rev. Med. Chem. 2013, 13, 814–823. [Google Scholar] [CrossRef]
- Ogawa, A. Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Molander, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 4, pp. 392–441. [Google Scholar]
- Tamai, T.; Nomoto, A.; Tsuchii, K.; Minamida, Y.; Mitamura, T.; Sonoda, M.; Ogawa, A. Highly selective perfluoroalkylchalcogenation of alkynes by the combination of iodoperfluoroalkanes and organic dichalcogenides upon photoirradiation. Tetrahedron 2012, 68, 10516–10522. [Google Scholar] [CrossRef]
- Kobiki, Y.; Kawaguchi, S.; Ogawa, A. Highly regioselective hydroselenation of inactivated terminal alkynes using diselenide–Ph2P(O)H mixed systems under visible-light irradiation. Tetrahedron Lett. 2013, 54, 5453–5456. [Google Scholar] [CrossRef]
- Tran, C.C.; Kawaguchi, S.-i.; Sato, F.; Nomoto, A.; Ogawa, A. Photoinduced Cyclizations of o-Diisocyanoarenes with Organic Diselenides and Thiols that Afford Chalcogenated Quinoxalines. J. Org. Chem. 2020, 85, 7258–7266. [Google Scholar] [CrossRef] [PubMed]
- Kobiki, Y.; Kawaguchi, S.-i.; Ohe, T.; Ogawa, A. Photoinduced synthesis of unsymmetrical diaryl selenides from triarylbismuthines and diaryl diselenides. Beilstein J. Org. Chem. 2013, 9, 1141–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kano, T.; Saito, S. Lead in Organic Synthesis. In Main Group Metals in Organic Synthesis; Yamamoto, H., Oshima, K., Eds.; Wiley-VCH: Weinheim, Germany, 2004; Volume 2, pp. 721–751. [Google Scholar]
- Elliott, G.I.; Konopelski, J.P. Arylation with organolead and organobismuth reagents. Tetrahedron 2001, 57, 5683–5705. [Google Scholar] [CrossRef]
- Finet, J.P.; Fedorov, A.Y.; Combes, S.; Boyer, G. Recent Advances in Ullmann Reaction: Copper(II) Diacetate Catalysed N-, O- and S- Arylation Involving Polycoordinate Heteroatomic Derivatives. Curr. Org. Chem. 2002, 6, 597–626. [Google Scholar] [CrossRef]
- Galingaert, G. The Organic Compounds of Lead. Chem. Rev. 1925, 2, 43–83. [Google Scholar] [CrossRef]
- Leeper, R.W.; Summers, L.; Gilman, H. Organolead Compounds. Chem. Rev. 1954, 54, 101–167. [Google Scholar] [CrossRef]
- Krause, E. Bleitriaryl, eine Parallele zum Triphenylmethyl; II.: Tricyclohexylblei. Chem. Ber. 1921, 54, 2060–2066. [Google Scholar] [CrossRef] [Green Version]
- Russell, G.A.; Tashtoush, H. Free-radical chain-substitution reactions of alkylmercury halides. J. Am. Chem. Soc. 1983, 105, 1398–1399. [Google Scholar] [CrossRef]
- Ogawa, A.; Takami, N.; Sekiguchi, M.; Yokoyama, H.; Kuniyasu, H.; Ryu, I.; Sonoda, N. A Novel Termal Addition of Diaryl Diselenide to Acetylenes. Chem. Lett. 1991, 20, 2241–2242. [Google Scholar] [CrossRef]
- Zhang, Q.-B.; Ban, Y.-L.; Yuan, P.-F.; Peng, S.-J.; Fang, J.-G.; Wu, L.-Z.; Liu, Q. Visible-light-mediated aerobic selenation of (hetero)arenes with diselenides. Green Chem. 2017, 19, 5559–5563. [Google Scholar] [CrossRef]
- Saba, S.; Rafique, J.; Franco, M.S.; Schneider, A.R.; Espíndola, L.; Silva, D.O.; Braga, A.L. Rose Bengal catalysed photo-induced selenylation of indoles, imidazoles and arenes: A metal free approach. Org. Biomol. Chem. 2018, 16, 880–885. [Google Scholar] [CrossRef]
- Rathore, V.; Kumar, S. Visible-light-induced metal and reagentfree oxidative coupling of sp2 C–H bonds with organo-dichalcogenides: Synthesis of 3-organochalcogenyl indoles. Green Chem. 2019, 21, 2670–2676. [Google Scholar] [CrossRef]
- Ye, Z.-P.; Xia, P.-J.; Liu, F.; Hu, Y.-Z.; Song, D.; Xiao, J.-A.; Huang, P.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Visible-Light-Induced, Catalyst-Free Radical Cross-Coupling Cyclization of N-Allylbromodifluoroacetamides with Disulfides or Diselenides. J. Org. Chem. 2020, 85, 5670–5682. [Google Scholar] [CrossRef]
- Dalberto, B.T.; Schneider, P.H. Photoinduced metal-free α-selenylation of ketones. RSC Adv. 2020, 10, 10502–10509. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.-Q.; Yi, W.; Wang, P.-F.; Liu, J.; Ma, M.; Hao, D.-Y.; Ming, L.; Ling, Y. Visible-light-induced oxidative coupling of vinylarenes with diselenides leading to α-aryl and α-alkyl selenomethyl ketones. Green Chem. 2021, 23, 1840–1846. [Google Scholar] [CrossRef]
- Rafique, J.; Rampon, D.S.; Azeredo, J.B.; Coelho, F.L.; Schneider, P.H.; Braga, A.L. Light-mediated Seleno-Functionalization of Organic Molecules: Recent Advances. Chem. Rec. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, N.; Dräger, M. Über gemischte Bindungen in der IV. Hauptgruppe, II Hexacyclohexylethan-Analoga Pb2(c-Hex)6, (c-Hex)3Pb—Sn(c-Hex)3 und Sn2(c-Hex)6. Z. Naturforsch. 1985, 40, 477–483. [Google Scholar] [CrossRef]
- Reich, H.J.; Renga, J.M.; Reich, I.L. Organoselenium Chemistry. Conversion of Ketones to Enones by Selenoxide Syn Elimination. J. Am. Chem. Soc. 1975, 97, 5434–5447. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Yang, H.; Fu, H. Visible-Light Photoredox Synthesis of Chiral α–Selenoamino Acids. Org. Lett. 2016, 18, 1968–1971. [Google Scholar] [CrossRef]
- Cohen, R.J.; Fox, D.L.; Salvatore, R.N. A Novel and Highly Efficient Synthetic Route to Unsymmetrical Organoselenides Using Cesium Bases. J. Org. Chem. 2004, 69, 4265–4268. [Google Scholar] [CrossRef]
- Duddeck, H.; Wagner, P.; Gegner, S. Dynamic 77Se NMR of phenylselenyl cyclohexane derivatives. Tetrahedron Lett. 1985, 26, 1205–1208. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | X | Light Source | Solvent | Temp., °C | Time, h | Yield, % 2 |
|---|---|---|---|---|---|---|
| 1 | S | white LED | CHCl3 | r.t. | 24 | 26 |
| 2 | S | xenon | CHCl3 | r.t. | 4 | 42 |
| 3 | S | dark | toluene | 100 | 12 | trace |
| 4 3 | S | dark | toluene | 100 | 12 | 13 |
| 5 | Se | white LED | CHCl3 | r.t. | 24 | 82 |
| 6 | Se | blue LED | CHCl3 | r.t. | 24 | 80 |
| 7 | Se | high-pressure Hg | CHCl3 | r.t. | 4 | 71 |
| 8 | Se | xenon | CHCl3 | r.t. | 4 | 93(87) |
| 9 | Se | xenon | C6H6 | r.t. | 4 | 88 |
| 10 | Se | xenon | MeCN | r.t. | 4 | 69 |
| 11 | Te | white LED | CHCl3 | r.t. | 24 | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, V.T.; Tran, C.C.; Yamamoto, Y.; Kodama, S.; Nomoto, A.; Ogawa, A. Clarification on the Reactivity of Diaryl Diselenides toward Hexacyclohexyldilead under Light. Molecules 2021, 26, 6265. https://doi.org/10.3390/molecules26206265
Hung VT, Tran CC, Yamamoto Y, Kodama S, Nomoto A, Ogawa A. Clarification on the Reactivity of Diaryl Diselenides toward Hexacyclohexyldilead under Light. Molecules. 2021; 26(20):6265. https://doi.org/10.3390/molecules26206265
Chicago/Turabian StyleHung, Vu Thai, Cong Chi Tran, Yuki Yamamoto, Shintaro Kodama, Akihiro Nomoto, and Akiya Ogawa. 2021. "Clarification on the Reactivity of Diaryl Diselenides toward Hexacyclohexyldilead under Light" Molecules 26, no. 20: 6265. https://doi.org/10.3390/molecules26206265
APA StyleHung, V. T., Tran, C. C., Yamamoto, Y., Kodama, S., Nomoto, A., & Ogawa, A. (2021). Clarification on the Reactivity of Diaryl Diselenides toward Hexacyclohexyldilead under Light. Molecules, 26(20), 6265. https://doi.org/10.3390/molecules26206265