1. Introduction
Natural marine polybrominated diphenyl ethers (PBDEs) are promising medicinal lead structures for anticancer [
1,
2] and antibacterial [
3] drugs. The latter can be rationalized by the structural resemblance to the man-made antibacterial, triclosan (2-(2′,4′-dicholorophenoxy)-5-chlorophenol). Structures of natural marine polybrominated products are derived from radical coupling of bromophenols catalyzed by a decarboxylative-halogenation enzyme [
4]. These structures can be classified into six types, characterized by the presence of 2-bromophenyl (7.9%), 4-bromophenyl (3.2%), 2,4-dibromophenyl (31.7%), 2-hydroxy-5-bromophenyl (3.2%), 2-hydroxy-3,5-dibromophenyl (36.5%), and 2-hydroxy-4,6-dibromophenyl moieties (1.6%), and other PBDEs, including dioxin, benzofuran type (15.9%). Therefore, over 60 natural marine PBDEs based upon these major frameworks have been discovered since 1969. Seven of them have been found in Indonesia [
5]. In addition, the unique structure of PBDEs, containing the H/C <1 (aromatic system), and the presence of halogen atoms are challenging for structure determination and correct assignments of
13C NMR chemical shifts. Currently, great improvements have made density functional theory (DFT)-based natural product structure determination a powerful method to provide accurate predictions and to evaluate
13C chemical shifts of target molecules possessing RMSE (root mean square error) <4.0 ppm [
6]. This method has been used to accurately evaluate conformationally flexible moieties [
6] and structures with high degree of unsaturation and substitution [
6,
7]. In general, the key features of computational NMR structure determination consist of conformational searches, DFT structure optimization, DFT energy calculations, NMR shift calculations, and statistical decisions. DFT calculations are also used to provide information about reactivity and kinetic stability of molecules, which may help us to understand the relationship between structures and their activities, as well as their quantum chemical parameters and properties, including frontier molecular orbitals (FMOs) and electronic charge distributions [
8,
9].
The distribution of natural marine PBDEs includes six phyla, of which the Porifera is the richest source. These molecules are most frequently found in the family
Dysideidae, especially in
Lamellodysidea herbacea and
Dysidea granulosa, which show different chemotaxonomic markers. Initial work on biosynthesis of PBDEs showed that they localize to sponge tissues inhabited by the symbiotic cyanobacterium,
Oscillatoria spongelliae [
10]. Recently, it was suggested that marine polybrominated diphenyl ethers are synthesized by symbiotic marine cyanobacteria (
Hormoscilla spongeliae or
Prochloron sp.) or by marine bacteria (
Pseudoaltromonas spp.) [
11,
12]. Despite the interesting discussion of PBDE origins in marine organisms, we re-investigated PBDEs from the abundant marine sponge,
L. herbacea collected from Indonesian waters, to determine their therapeutic potential as anticancer or antibacterial drugs, by chemical reactions and computational studies. The Gram-positive,
Staphyloccoccus aureus and Gram-negative
Klebisella pneumoniae, are two important pathogenic targets for searching new antibiotics known as ESKAPE pathogens (
Enterococcus faecium,
Staphylococcus aureus,
Klebsiella pneumoniae,
Acinetobacter baumanii,
Pseudomonas aeruginosa and
Enterobacter species [
13]. Moreover, new antibiotic scaffolds for Gram-negative pathogens have been extremely rare in recent decades.
The major PBDE
1 was isolated in gram quantities, having the largest number of substituted bromine atoms in two phenol rings. Its structure was determined with X-ray analysis [
14]. PBDE
1 is a signature compound of
L. herbacea collected in Indonesia and is frequently found in high quantities. We observed that the biological activity of PBDE molecules depends on two functional characteristics: The presence of phenol and the number and position of bromine atoms [
14,
15]. Starting with
1 (2, 3, 4, 5-tetrabromo-6-(3ʹ, 5ʹ-dibromo-2ʹ-hydroxyphenoxy) phenol), debromination via the reversibility of electrophilic aromatic bromination with regioselective debromination [
16,
17] may be a good way to synthesize various PBDE molecules with a lower number and a variety of positions of bromine atoms, in addition to isolation of other analogues and modifications of phenolic functional groups by methylation and acetylation. With the foregoing background, this article describes isolation of natural PBDE analogues (
1–
6) from the sponge,
L. herbacea including evaluation of their
13C NMR chemical shifts using an advanced computational method [
6]. It also reports structural modifications involving transformation of phenolic functional groups by methylation and acetylation to give seven molecules (
7–
13) and an interesting debromination in the presence of a bromine scavenger to give nine additional molecules (
1,
2,
14,
16–
18,
20,
23,
26) (
Figure 1). We propose the calculated
13C NMR data of 30 PBDEs and evaluate their RMSEs for which we have experimental data. In regard to the exploration reaction, we also found that acetylation occurred in the presence of Ac
2O and sonication without a catalyst and without solvent at room temperature for 2–3 h. Two compounds (
8 and
9) have new crystal structures, whereas compounds
10,
12, and
13 are entirely new structures. Compound
18 is a new derivative obtained via debromination. One unpublished
13C NMR of
6 is also reported and secured its assignment by its calculated
13C NMR chemical shifts. This article also describes structure-activity relationships of marine polybrominated natural products against human embryonic kidney (HEK293T) cells and Gram-positive
Staphylococcus aureus, as well as the Gram-negative bacterium
Klebsiella pneumoniae. Computational studies further support their activities and account for acetylation and debromination.
2. Results and Discussion
Six known O-PBDEs (
1–
6) have been isolated from the Indonesian marine sponge,
L.
herbacea, collected at Ujung Kulon. MS and
1H NMR spectra of
1−
6 were in good agreement with previously reported data [
14,
15,
18,
19,
20]. Compound
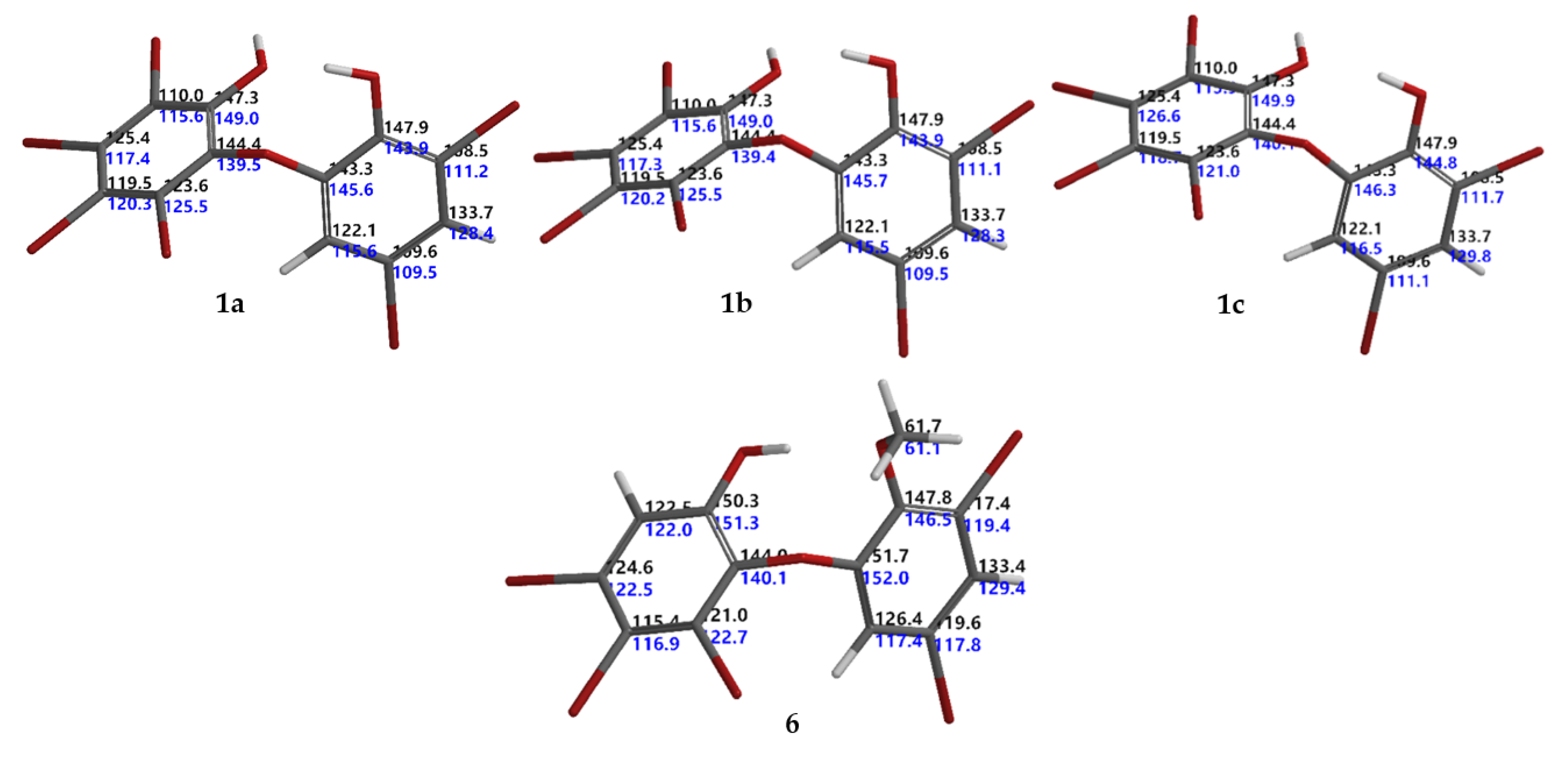
6 was first isolated by Fu et al., in 1995 [
20]. To the best of our knowledge,
13C NMR data for compound
6 have not been reported previously. This work also gave us an opportunity to assign the
13C NMR for compound
6 (
Table 1). Complete
13C NMR data of
6 (HREIMS
m/z 605.6311 [M]
+, C
13H
7O
379Br
5, Δ −0.0001 mmu) were derived with the aid of DEPT, HMQC, and HMBC experiments and analogies from the literature. The HMBC experiment showed correlations with H2/C1,3,4,6; H4′/C2′, 3′, 5′, 6′; H6′/C1′,2′, 3′, 5′ and –OMe/C2′ leading to the order of the
13C data of
6 (
Table 1). Moreover, the assignment was secured by the protocol of Hehre et al. [
6] to give RMSE < 4.0 ppm (RMSE obtained was 3.2 ppm; RMSE for aromatic compounds were around 3.2 [
6]). Intriguing with the RMSE of the isolated natural products, we conducted a set of experiments in order to obtain theoretical
13C NMR chemical shifts of
1–
5 with six steps: (1) conformational search using the MMFF molecular mechanics model; (2) calculation of equilibrium geometries using the HF/3-21G model; (3) calculation of energies using the ωB97X-D/6-31G* density functional model; (4) calculation of equilibrium geometries using the ωB97X-D/6-31G* density functional model; (5) calculation of energies of using the ωB97X-V/6-311+G(2df,2p)(6-311G*) density functional model; and (6) calculation of
13C NMR chemical shifts using the ωB97X-D/6-31G* density functional model, correction of
13C NMR chemical shifts based on the empirical parameters, and correction
13C NMR chemical shifts based on the Boltzmann weight obtained in step 5. We found that compounds
2–
6 gave RMSEs <4.0 ppm, except for
1 (RMSE >4.0 ppm, for
1a 4.3 ppm [
18], for
1b 4.4 ppm [
21] in DMSO-
d6). These values suggested a possible error in the
13C chemical assignments. The error could be in an assignment of
13C chemical shifts in ring A (lack of H atoms). Based on the calculated
13C chemical shifts, we propose an RMSE for
1 as 3.5 ppm as in
1c (Me
2CO-
d6). The assignment of
13C chemical shifts for
1 and
6 can be seen in
Figure 2.
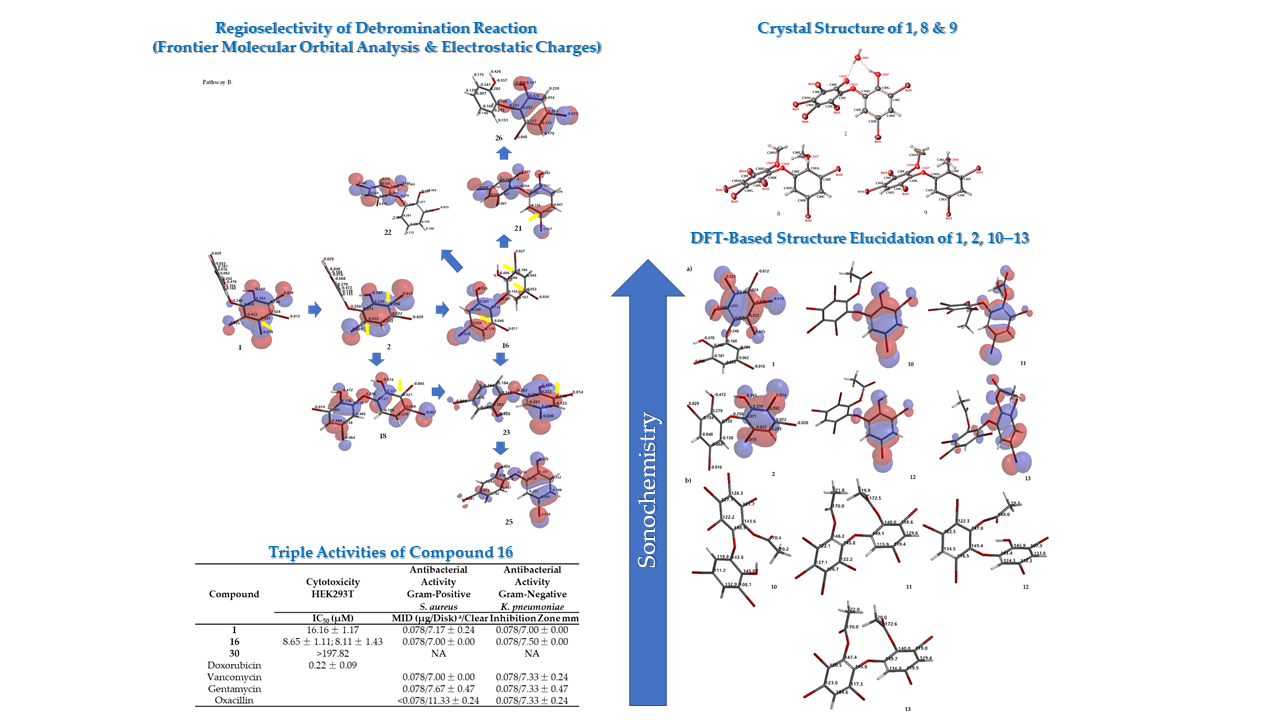
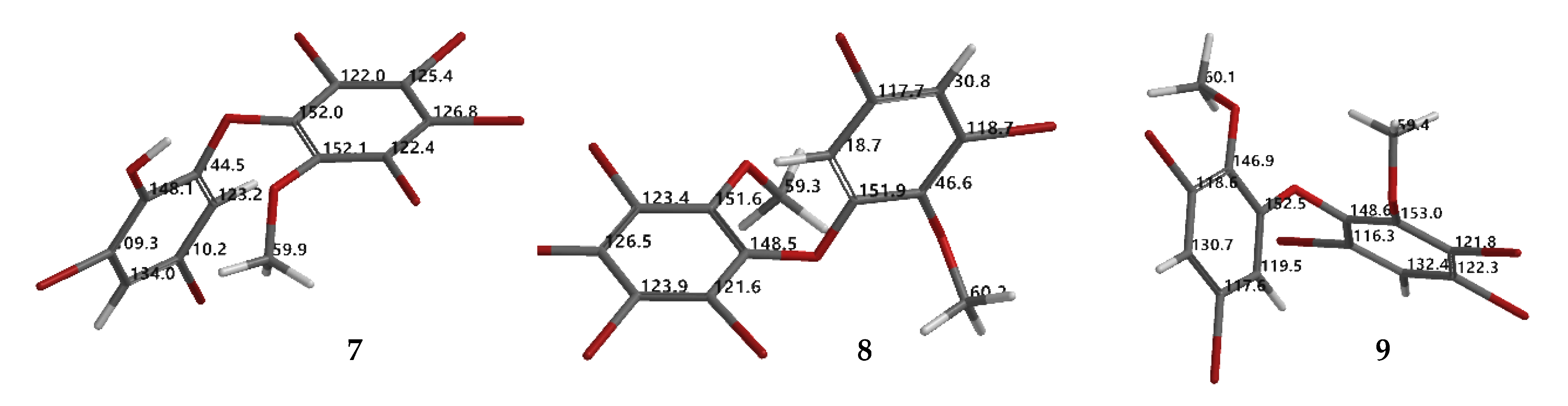
In order to see the effect of phenol in our assays, seven-modified phenolic compounds (7–13) were prepared by methylation (7–9) and acetylation (10–13). Two new crystal structures (8 and 9) were obtained from a methylation reaction using TMSCHN2 with three new synthetic PBDE derivatives (10, 12, 13) from an acetylation reaction using green chemistry.
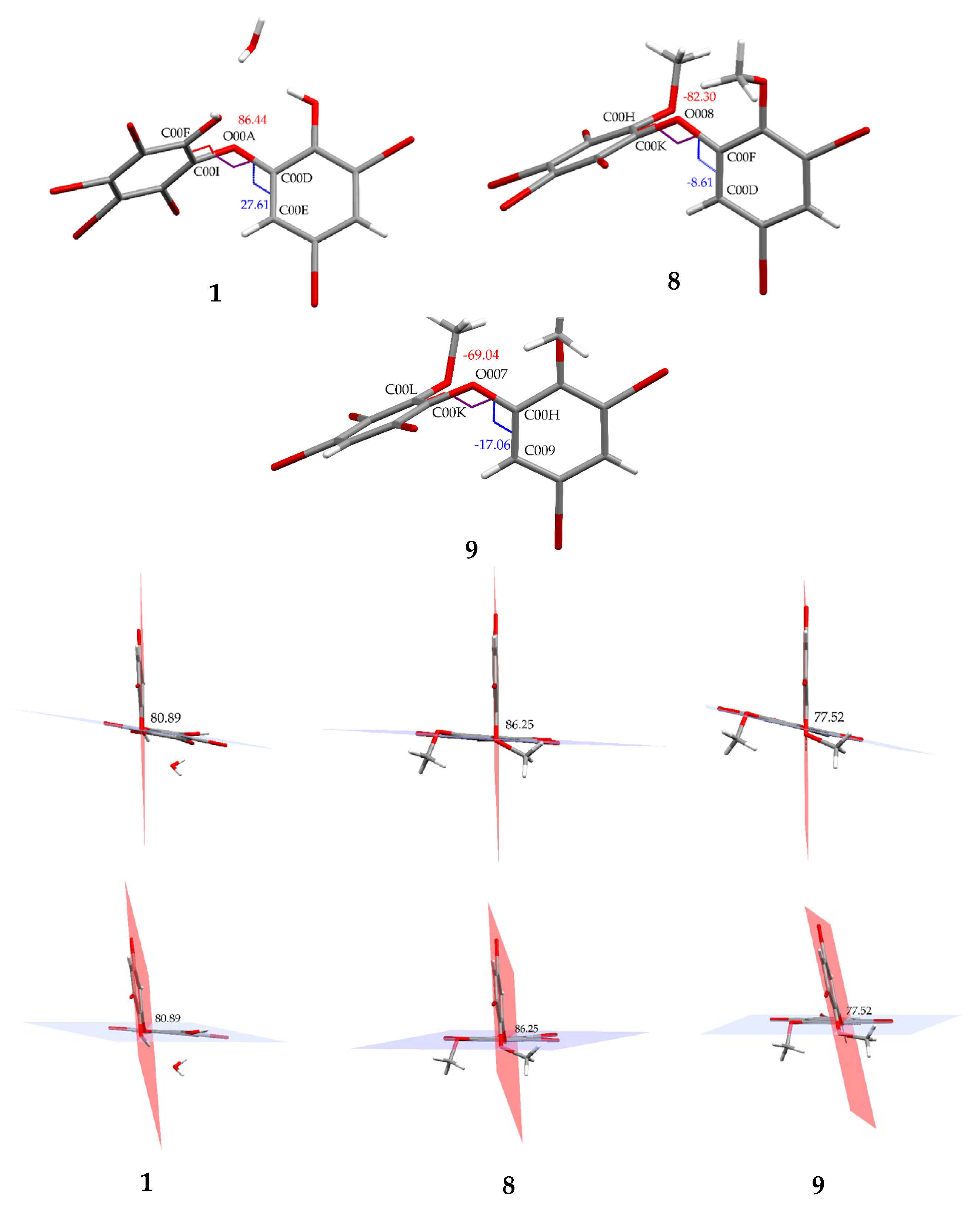
Compound 8 (C14H8Br6O3) is 2,3,4,5-tetrabromo-6-(3′,5′-dibromo-2′-methoxyphenoxy) anisole, which has torsion angle φ1 = −8.6° (C6′– C1′–O–C6); φ2 = −82.3° (C1′–O–C6–C1), while the ether bond and dihedral angle between two aromatics are 116.9 (4)° and 86.3°, respectively. Compound 8 was crystallized in the triclinic space group P-1 (Z = 2) using CHCl3−MeCN (3:2).
Compound
9 (C
14H
9Br
5O
3) was identified as 2,3,5-tribromo-6-(3′,5′-dibromo-2′-methoxyphenoxy) anisole after X-ray analysis. The torsion angle of
9 is
φ1 = −17.1° (C6′– C1′–O–C6);
φ2 = −69.0° (C1′–O–C6–C1), whereas the rest are 117.3 (6)° and 77.5°. Compound
9 was crystallized as orthorhombic in the space group Pca21 using CHCl
3−Me
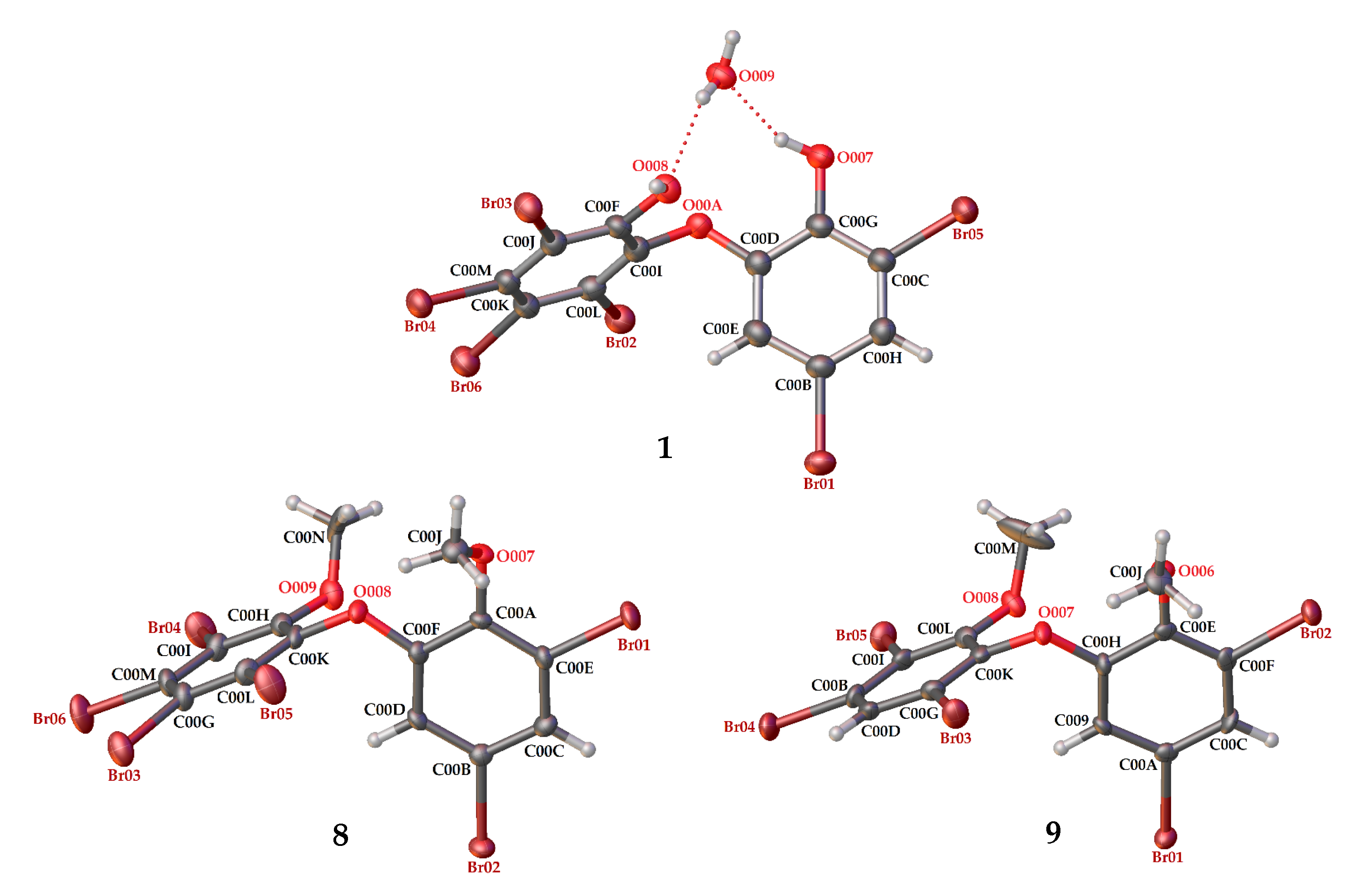
2CO−MeCN (1:1:1). Crystal structures of
1,
8 and
9 are depicted in
Figure 3 and
Figure 4, while theoretical
13C NMR chemical shifts of
7–
9 are shown in
Figure 5. The RMSE of
7 showed 3.8 ppm after comparing with the literature data [
19].
We initially screened conditions for acetylation and found that
1 reacts with Ac
2O under sonication for 1 h without DMAP and Et
3N at room temperature. Conversion of
1 to
11 was estimated at >95% using TLC. A larger amount of
1 was used under the above conditions to give acetylation products (
10–
13) with slightly longer time (2–3 h). To date, acetylation with acyl halides or acid anhydrides has been reported using solvents and catalysts at room temperature or higher (>40 °C). Acetylation methods have been reported without catalysts at high solvent temperatures (60–70 °C) or with a variety of catalysts from room temperature to 110 °C [
22]. With regard to PBDE molecules, only
11 has been obtained from acetylation using Ac
2O with pyridine at room temperature for 24 h [
23]. Therefore, our method is new and is consistent with green chemistry concepts.
Compound
10 is a partial acetylation product (
Rf 0.23 Hex/EtOAc 8:1, n-silica, UV λ 254 nm) of
1, based on its HRESIMS [M + H]
+ 716.5404 (C
14H
7O
479Br
481Br
2, Δ 0 mmu).
1H NMR data confirmed the presence of one -OAc group by observing
δ (ppm) 2.47 in CDCl
3 or 2.40 in Me
2CO-
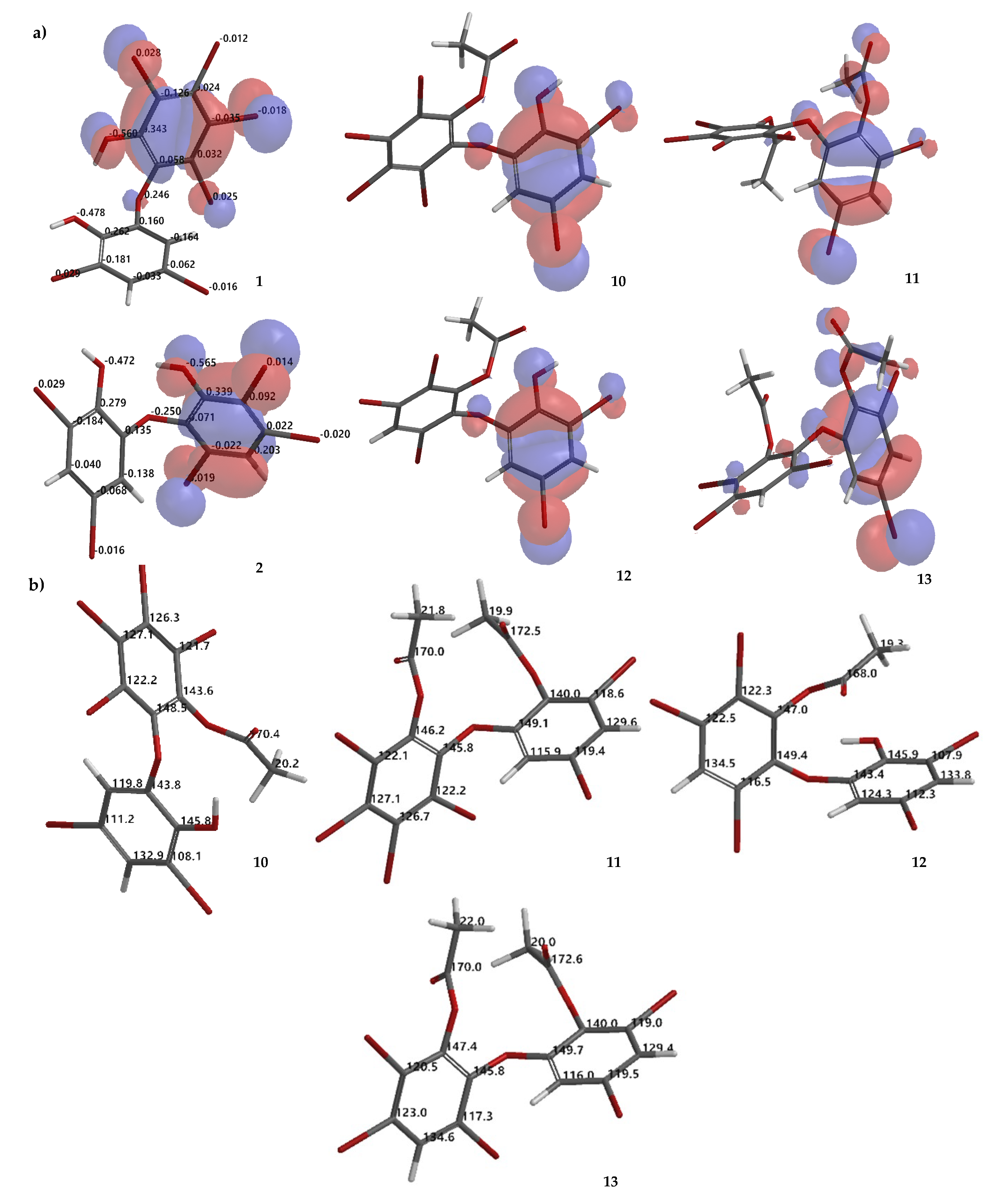
d6. Because ring A is fully substituted, no HMBC correlation can be observed. The -OAc group can be placed on ring A by assuming that ring A is more reactive than ring B, on the basis of molecular orbital analysis of
1 with DFT dispersion correction level of theory and a triple-ζ-basis set (
Figure 6a). Moreover, analysis of the electrostatic charge of
1 also justified that the -OH group in ring A is more electronegative (−0.560) than that of ring B (−0.478) (
Figure 6a). Therefore, the first acetylation is more likely to proceed in ring A. In addition, comparison of
10 with
3 and
7 containing an anisole group on ring A showed that the
1H chemical shift is more upfield than ring B. Compound
10 was revealed as 2,3,4,5-tetrabromo-6-(3′,5′-dibromo-2′-hydroxyphenoxy) phenyl acetate.
Compound
11 is 2,3,4,5-tetrabromo-6-(3′,5′-dibromo-2′-acetoxyphenoxy) phenyl acetate, possessing
Rf 0.52 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm) and HRESIMS [M + Na]
+ 780.5329 (C
16H
8O
579Br
481Br
2Na, Δ −0.0002 mmu) and after comparing its
1H NMR data with the literature [
23]. The
1H chemical shift of the-OAc group in ring A could be assigned more upfield by comparing these data with the
1H NMR of methylated products, as in
8 and
9. Therefore,
δ 2.24 could be placed on ring A, while
δ 2.36 (Me
2CO-
d6) could be assigned on ring B.
Compound
12 (
Rf 0.34 Hex/EtOAc 8:1, n-silica, UV λ 254 nm) is a mono-acetylated product of
2, evidenced by HRESIMS [M + H]
+ 640.6278 (C
14H
8O
479Br
281Br
3, Δ 0 mmu). The
1H chemical shift of the -OAc was observed at
δ (ppm) 2.47 in CDCl
3 or 2.40 in Me
2CO-
d6. The same situation as
10 was also observed for
12. The -OAc group could be attached to ring A. Highest occupied molecular orbital (HOMO) analysis
2 showed that ring A is more reactive than ring B which is justified by electrostatic charge analysis of the -OH group in ring A (−0.565) and in ring B (−0.472) (
Figure 6a). Compound
12 was confirmed as 2,3,5-tribromo-6-(3′, 5′-dibromo-2′-hydroxyphenoxy) phenyl acetate.
Compound
13 (
Rf 0.51 Hex/EtOAc 8:1, n-silica, UV λ 254 nm) is a completely acetylated product of
2. Two attached -OAc groups were verified with HRESIMS [M + Na]
+ 704.6226 (C
16H
9O
579Br
281Br
3Na, Δ 0.0023 mmu).
1H chemical shifts of two -OAc groups could be observed at
δ (ppm) 2.25 and 2.36 in CDCl
3 or 2.24 and 2.36 in Me
2CO-
d6. The -OAc group on ring A could be
δ 2.24, while the other -OAc could assigned to
δ 2.36 (in Me
2CO-
d6). Compound
13 is determined as 2,3,5-tribromo-6-(3′,5′-dibromo-2′-acetoxyphenoxy) phenyl acetate. To complete the characterization of acetylation products
10–
13, theoretical
13C NMR chemical shifts of the compounds are proposed as in
Figure 6b.
Next, we examined the effect of lower bromine atoms on two phenol rings by employing debromination. Compound
1, isolated in gram quantities, and possessing six bromine atoms on two phenol rings, was subjected to refluxing using HBr and Na
2SO
3 as scavengers of bromine in the presence of acetic acid. Compounds (
1,
2,
14,
16–
18,
20,
23,
26) were isolated and confirmed from the reaction. MS and
1H NMR spectra of compounds (
1,
2,
16,
17,
20,
23, 26) were in a good agreement with previous reports [
14,
15].
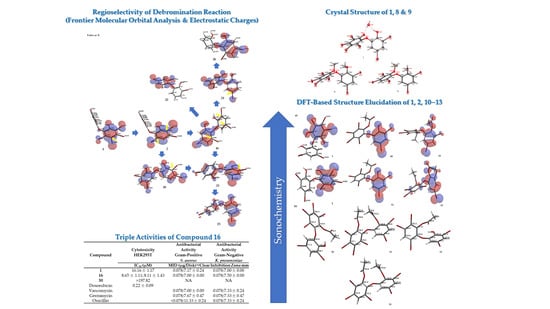
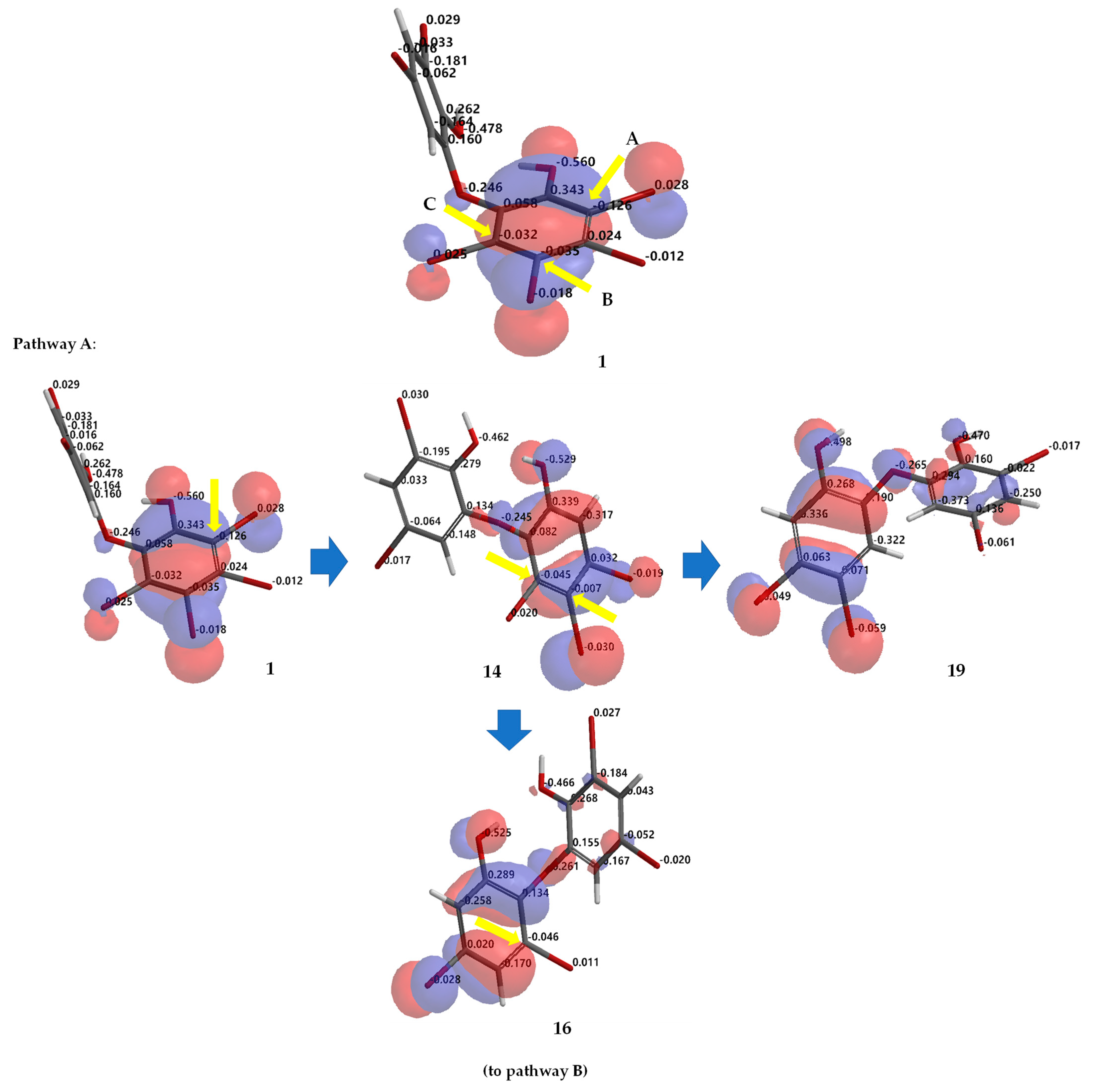
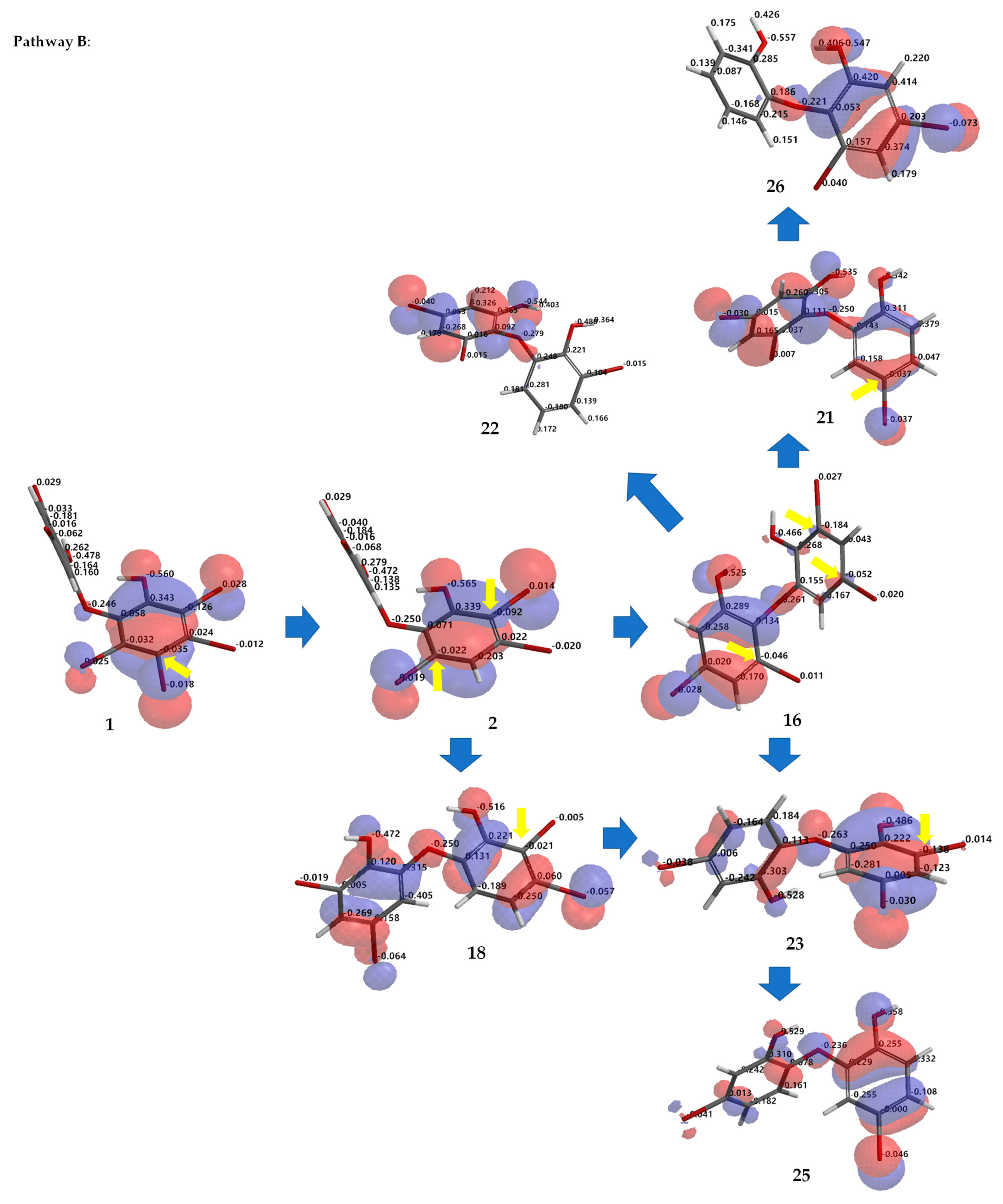
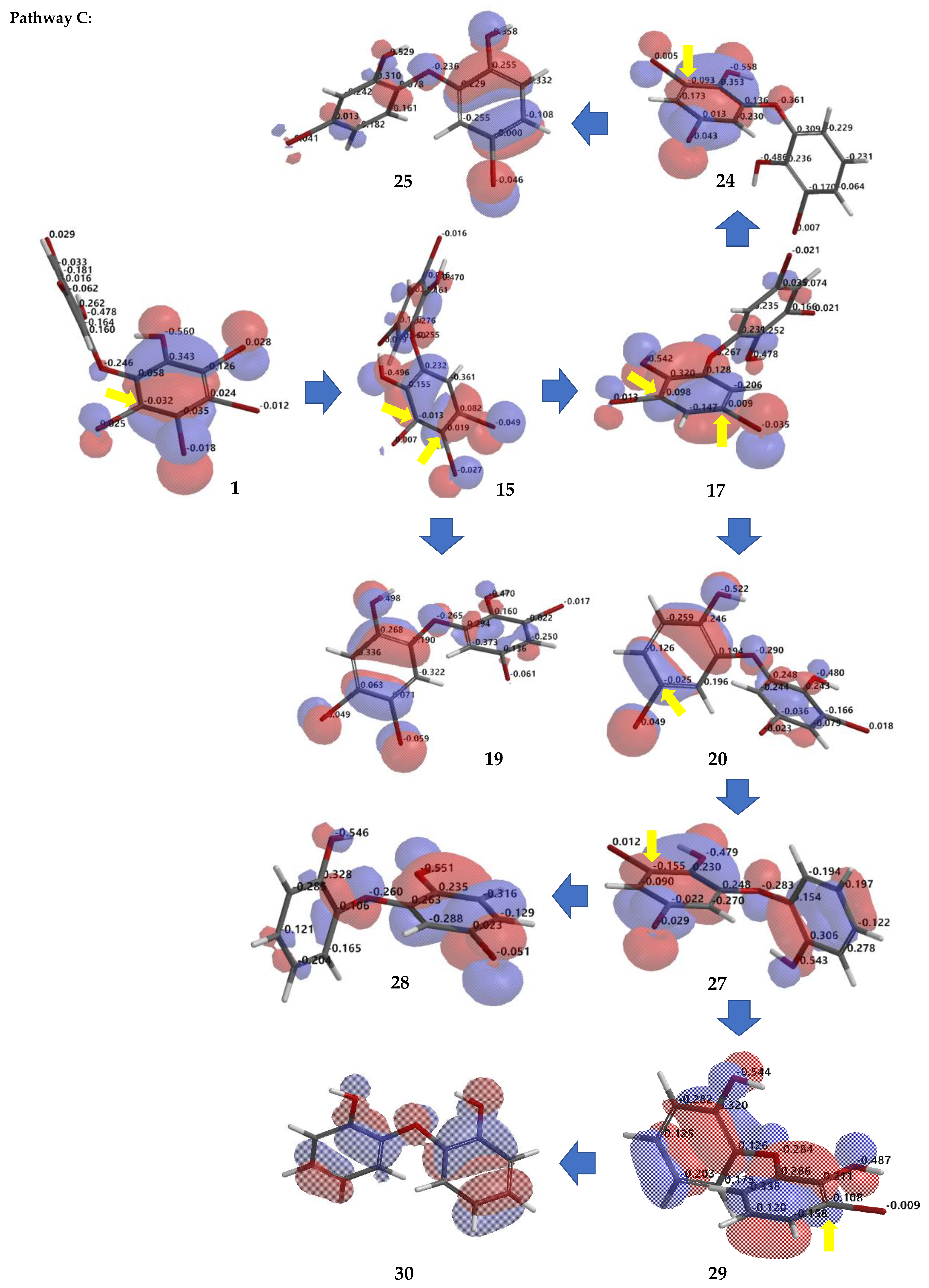
Debromination of
1, including its product, can be supported by DFT ωB97X-D/6–311 + G(2d,p) calculations. Combination analysis of frontier molecular orbital (HOMO) for
1,
2,
14,
16–
18,
20,
23, 26 and their electrostatic charge are shown in
Figure 7 allowing us to propose the three putative debromination pathways.
Only
o,
p-bromines were selectively reduced; hence, we could deduce the isolated products with the three putative debromination pathways
A–
C shown in
Figure 7. Debromination in this study gave nine compounds, (
1,
2,
14,
16–
18,
20,
23, 26). In addition, there were
8 hypothetical compounds (
21,
22,
24,
25,
27–
30) that we have predicted on the basis of computational studies. This includes
15, which was reported previously [
21,
24,
25]. Based on analysis of FMO and electrostatic charge for
1 on ring A, three reactive sites were readily identified in HOMO region as C2 (electrostatic charge: −0.125), C5 (−0.035), and C6 (−0.032) corresponding to Br atoms in the
ortho and
para positions toward the -OH group and the Br atom in the
ortho position toward the −OR group, respectively which led to the three putative pathways
A–
C. Moreover, combination analysis of FMOs and electrostatic charges for the three pathways support the presence of debromination products as in
1,
2,
14,
16–
18,
20,
23, and
26. The high electron density and negative charge gave the possibility of attack through the
ortho or
para positions of the ring toward the lowest unoccupied molecular orbital (LUMO) HBr orbital leading to the selectivity of the reaction. Reaction products were predicted to occur via the
ortho and
para pathways toward hydroxy or phenoxy groups. This was because of competition between two activating groups (regioselective). Reaction products predicted to occur via the
ortho toward hydroxy groups (pathway
A) were
1 →
14 →
16 (the scheme continues on the pathway
B);
1 14 →
19. Reaction products predicted to occur via the
para toward hydroxy groups (pathway
B) were
1 →
2 →
16 →
21 →
26;
1 →
2 →
16 →
22;
1 →
2 →
16 →
23 →
25;
1 →
2 →
18 →
23 →
25. Reaction products predicted to occur via the
para toward phenoxy groups (pathway
C) were
1 →
15 →
17 →
20 →
27 →
28; 1 →
15 →
17 →
20 →
27 →
29 →
30;
1 →
15 →
17 →
24;
1 →
15 →
19.
Based on
Figure 7, we also observed the possibility of isomerization, which also supports the debromination mechanism proposed by Effenberger [
17]. This may explain other products that are not directly obtained by removing bromine atoms in the
ortho or
para positions, to the electron-donating groups (EDG) -OH or-OR. Among the debromination products, compound
26 was the most stable (ΔE
LUMO-HOMO 9.38 eV) followed by
16 (9.14 eV), while less stable compound was
1 (8.62 eV). All hypothetical compounds (
21,
22,
24,
25,
27–
30) were relatively stable (9.03–9.29 eV) except for
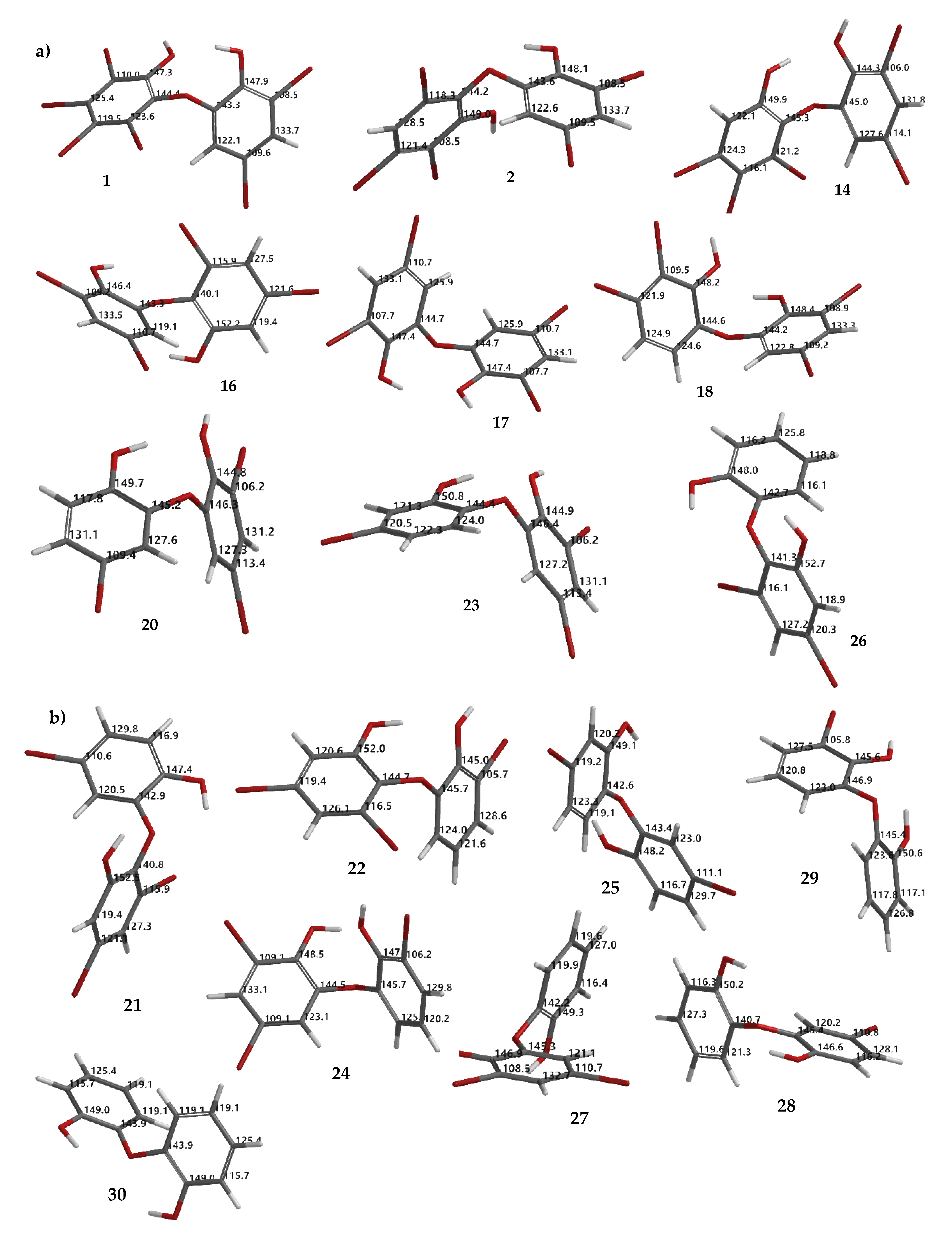
24 (8.86 eV). Apparently, the molecules may exist during the reaction, offering a challenging task to isolate and to assay them with our interest assay panel because of nature of the molecules. In this computational study, we also proposed theoretical
13C NMR data for the experimental (
1,
2,
14,
16–
18,
20,
23, and
26) (
Figure 8a) and hypothetical (
21,
22,
24,
25,
27–
30) (
Figure 8b) molecules.
A series of PBDE molecules was evaluated for cytotoxicity using human embryonic kidney (HEK293T) cells, Gram-positive
Staphylococcus aureus, and Gram-negative
Klebsiella pneumoniae. Compound
1 (2, 3, 4, 5-tetrabromo-6-(3′, 5′-dibromo-2′-hydroxyphenoxy) phenol) showed cytotoxicity against HEK293T cells at IC
50 16.16 ± 1.68 µM, whereas compound
16 (3, 5-dibromo-6-(3′, 5′-dibromo-2′-hydroxyphenoxy) phenol) was checked twice against HEK293T cells from different reactions, showing IC
50 8.65 ± 1.11 and IC
50 8.11 ± 1.43 µM, respectively. Compound
30 (2, 2′-oxydiphenol) showed no cytotoxicity against HEK293T cells with an IC
50 >197.82 µM. This result suggests that four bromines and their positions are important for cytotoxicity against HEK293T cells, as in
16. Moreover, both bacteria showed MID = 0.078 µg/disk for compounds
1 and
16 (
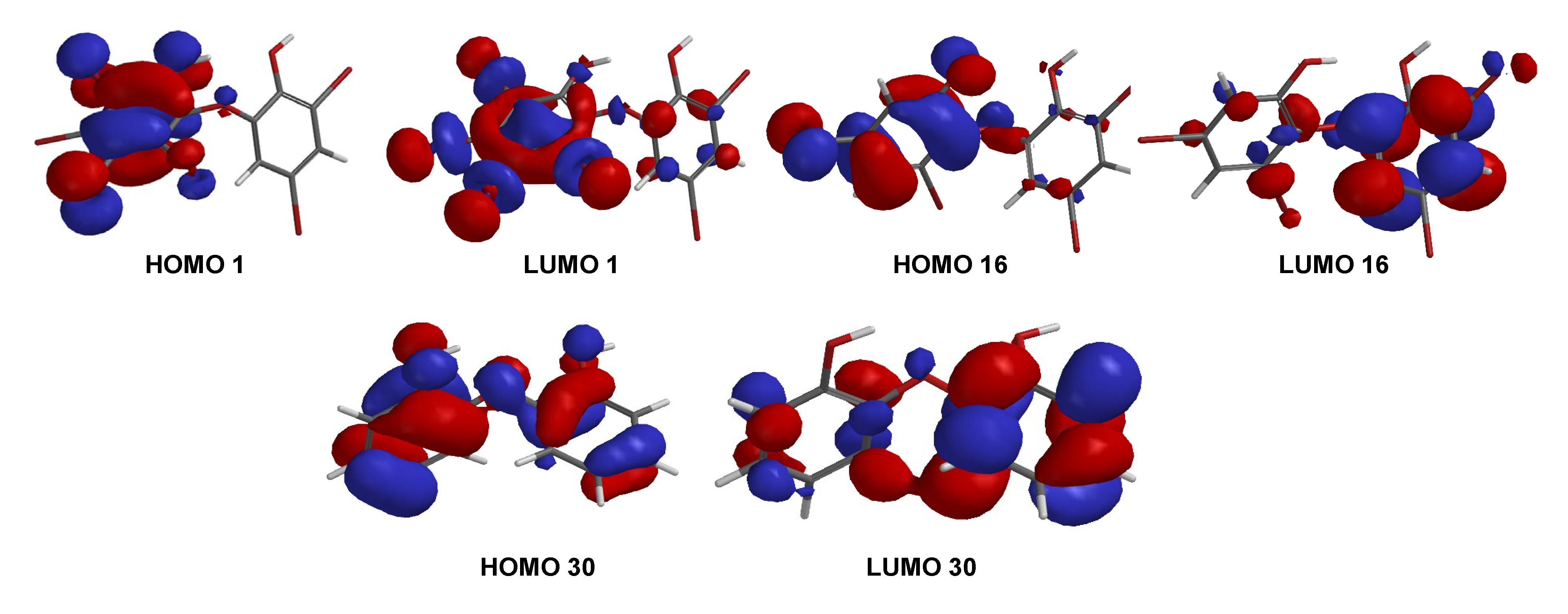
Table 2). The result was supported by the shape of HOMO and LUMO for
1,
16, and
30 (
Figure 9). Compounds
1 and
16 showed the HOMO–LUMO orbitals located in different molecular regions and this characteristic may explain their potent antibacterial activity, as shown in many studies [
9,
26,
27]. In contrast,
30 had HOMO–LUMO orbitals dispersed across almost the entire molecular region. While the MID of
1 and
16 were the same, the clear inhibition zone of
1 is slightly stronger than that of
16. This was supported by E
LUMO of
1 (−0.05 eV) < E
LUMO of
16 (0.38 eV) while E
LUMO of
30 was 1.13 eV. It is clear that the higher number of bromine atoms, as in
1 stabilizes LUMO energy and gives slightly stronger antibacterial activity against the Gram-positive bacterium,
S. aureus [
27]. In contrast, the antibacterial activity of
16 against Gram-negative bacterium,
K. pneumoniae, was slightly more potent than that of
1.
Compound
16 was discovered previously in the marine sponge,
Dysidea herbacea collected from Australia [
29] and later in
Lamellodysidea sp. [
24] collected from Papua New Guinea. It showed a wide range of antibiotic activities against
Staphylococcus aureus (MIC 1.25–1.6 µg/mL),
Enterococcus faecium (MIC 1.6–3.1 µg/mL),
Escherichia coli (MIC 50 µg/mL),
Pseudomonas aeruginosa and
Candida albicans (MIC >50 µg/mL), while it also had an IC
50 >50 µg/mL against Bsc-1 cells [
24]. Methylation and acetylation of
1 reduced cytotoxicity, with an IC
50 >10 µg/mL against HEK293T, as in
7–
13. Meanwhile, antibacterial activity of methylation and acetylation products was weaker, with MID >0.078 µg/disk against Gram-positive
Staphylococcus aureus and Gram-negative
Klebsiella pneumoniae.
In summary, we found new derivatives
10,
12,
13 from acetylation by comparing data published between 1969 and 2020. We performed the acetylation with a new method using green chemistry at room temperature and determined the structures using analysis of FMOs and electrostatic charges. In addition, we also discovered a new debromination product
18 and new crystal structures of methylated
8 and
9 with twist conformations (
φ1,
φ2 > 0°) [
30]. The presence of debromination products (
1,
2,
14,
16–
18,
20,
23,
26) and hypothetical molecules (
21,
22,
24,
25,
27–
30) were predicted using analysis of FMOs and electrostatic charges showing regioselectivity
ortho and
para positions toward the EDG -OH or -OR groups in PBDEs. The analysis also showed the possibility of isomerization among the PBDEs. All products methylations, acetylations, and debrominations were characterized for their theoretical
13C NMR chemical shifts. Novel
13C NMR data
6 are also reported (RMSE 3.2 ppm). PBDE compounds that lose hydroxyl groups, due to methylation and acetylation, have weaker biological activity. Fewer bromines, as in
16, resulted in a significant IC
50 8.65 ± 1.11; 8.11 ± 1.43 µM against HEK293T cells. Compound
16 also showed antibacterial activity against Gram-positive
Staphylococcus aureus, as well as Gram-negative
Klebsiella pneumoniae, with an MID = 0.078 µg/disk. Cytotoxicity and antibacterial assays of derived compounds show that two phenolic hydroxyl groups and four bromine atoms are important for these activities. The result of active compounds was supported by analysis of FMOs. Additional information can be found in the
Supplementary Materials.
3. Materials and Methods
3.1. General Methods
NMR spectra were measured on a 500 MHz Bruker Avance III spectrometer (MA, USA) or a 500 MHz JEOL (Tokyo, Japan) or a 500 MHz Varian (CA, USA). Chemical shifts were referenced to tetramethylsilane (TMS) or acetone (Me2CO) signals. MS spectra were recorded on a Waters Acquity Xevo G2-S ESIQTOF in positive mode or an HRESITOFMS JEOL T100LP, or EIMS were measured on a Hitachi M-2500 instrument. UV and IR spectra were obtained on a Perkin Elmer Spectrum One FTIR and on a Shimadzu Pharmaspec 1700 spectrophotometer. X-ray analysis was performed on a Rigaku AFC10 goniometer equipped with a Saturn 724+ detector. High-performance liquid chromatography (HPLC) separations were carried out on a Hitachi L-6000 pump fitted with Shodex RI-101 refractive index and SPD-20A Shimadzu UV detectors, or a Shimadzu HPLC with Prominence LC-20AD, DGU-20A5, SPD-20A. A Cosmosil 5SL-II-MS (10 × 250 mm) column was used for HPLC. Analytical thin-layer chromatography (TLC) was performed on Merck silica gel 60 F254 plates and visualized with sulfuric acid with cerium sulfate. All solvents used were reagent grade.
3.2. Animal Material
Marine sponges were collected by hand while scuba diving in Banten Province, Indonesia at a depth of 5–10 m. Samples were then stored in EtOH. Sponges were identified as Lamellodysidea herbacea by NJdeV.
3.3. Extraction and Isolation
A fresh marine sponge specimen (wet weight 300 g) stored in EtOH was extracted with MeOH. The combined extract was concentrated under vacuum, and the resulting residue was partitioned between hexane and aqueous MeOH (90%). The latter layer was further partitioned between CH2Cl2 and aqueous MeOH (50%). Finally, the aqueous MeOH (50%) was removed and adjusted with water, followed by extraction with n-BuOH. The three layers: Hexane, CH2Cl2 and BuOH layer were evaluated for activity against Gram-positive and Gram-negative bacteria. Recrystallization of the CH2Cl2 layer using CHCl3–Me2CO–MeCN gave 1 (1.36 g). A non-crystalline fraction was separated using either a silica gel column or silica HPLC eluted with hexane/EtOAc/MeOH, followed by recrystallization to give compound 2 (5.4 mg), a mixture of compounds 2 and 3 (5.6 mg), compound 4 (7.3 mg), and compound 5 (0.8 mg). Additional 2 (8.3 mg) and 3 (11.9 mg) were isolated from the hexane fraction after open column chromatography. Compounds 4 (163.5 mg) and 6 (50.6 mg) were isolated from the hexane layer collected from another L. herbacea.
3.4. Methylation
To a solution of 1 (9.1 mg) in MeOH (1 mL), 2 M TMSCHN2 in hexane was added dropwise. The reaction was monitored by TLC. The solution was allowed to stand at room temperature and concentrated to dryness under a stream of nitrogen followed by purification using HPLC (RP 18, MeOH, MeCN + 0.1% TFA) to give 7 (3 mg) and 8 (2.4 mg). Compound 7: Rf 0.38 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 3.87 (3H, s), 6.88 (1H, d, J = 2.2 Hz), 7.43 (1H, d, J = 2.2 Hz), HRESIMS m/z 690.0626 [M + H]+ (C13H6O379Br581Br, Δ −0.4808 mmu). Compound 8: Rf 0.57 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm), 1H NMR (CDCl3) δ 3.85 (3H, s), 4.00 (3H, s), 6.97 (1H, d, J = 2.2 Hz), 7.50 (1H, d, J = 2.2 Hz), HRESIMS m/z 702.6289 [M + H]+ (C14H9O379Br481Br2, Δ 0.0678 mmu).
To a solution of 2 (5.2 mg) in MeOH (1 mL) excess 2 M TMSCHN2 in hexane was added and monitored by TLC. The solution was allowed to stand at room temperature and concentrated to dryness under a stream of nitrogen to give the total methyl derivative 9 (5.2 mg). Compound 9: Rf 0.63 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm), 1H NMR (CDCl3) δ 3.82 (3H, s), 4.00 (3H, s), 6.50 (1H, d, J = 2.2 Hz), 7.40 (1H, d, J = 2.2 Hz), 7.76 (1H, s), HRESIMS m/z 630.5989 [M + H]+ (C14H10O381Br5 Δ −0.0456 mmu).
A solution of 4 (8.9 mg) in MeOH (1 mL) was treated similarly with diluted TMSCHN2 to give compound 9 (8.9 mg). Compound 9: Rf 0.63 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm, 1H NMR (CDCl3) δ 3.82 (3H, s), 4.00 (3H, s), 6.50 (1H, d, J = 2.2 Hz), 7.40 (1H, d, J = 2.2 Hz), 7.76 (1H, s), HRESIMS m/z 630.5989 [M + H]+ (C14H10O381Br5 Δ −0.0456 mmu).
3.5. Acetylation
Screening of acetylation was performed under seven conditions with a variety of catalysts and solvents, without catalyst and solvent, with or without sonication. Acetylation of 1 with Ac2O and sonication for 1 h proceeded to give 10 and 11. Acetylation of 2 with Ac2O and sonication for 1 h proceeded to give 12 and 13. The reaction was performed without DMAP, Et3N, and solvent.
To 1 (15 mg) Ac2O (2.1 mmol) was added and sonicated for 2–2.5 h at room temperature to give 10 (1.1 mg) and 11 (6.4 mg) after purification by HPLC (RP18, MeCN + 0.1% TFA). Compound 10: Rf 0.23 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 2.40 (3H, s), 7.12 (1H, d, J = 2.2 Hz), 7.62 (1H, d, J = 2.2 Hz), 1H NMR (CDCl3) δ 2.47 (3H, s), 6.62 (1H, d, J = 2.1 Hz), 7.51 (1H, d, J = 2.1 Hz), HRESIMS m/z 716.5404 [M + H]+ (C14H7O479Br481Br2, Δ 0 mmu). Compound 11: Rf 0.23 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 2.24 (3H, s), 2.36 (3H, s), 7.14 (1H, d, J = 2.1 Hz), 7.64 (1H, d, J = 2.1 Hz), HRESIMS m/z 780.5329 [M + Na]+ (C16H8O579Br481Br2Na, Δ −0.0002 mmu).
To 2 (15.3 mg) Ac2O (2.1 mmol) was added and sonicated for 2–2.5 h at room temperature to give 12 (3.7 mg) and 13 (2.2 mg) after purification using HPLC (RP 18, MeCN + 0.1% TFA). Compound 12: Rf 0.34 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 2.40 (3H, s), 7.00 (1H, d, J = 2.1 Hz), 7.62 (1H, d, J = 2.1 Hz), 7.66 (1H, s), 1H NMR (CDCl3) δ 2.47 (3H, s), 6.63 (1H, d, J = 2.1 Hz), 7.50 (1H, d, J = 2.1 Hz), 7.54 (1H, s), HRESIMS m/z 640.6278 [M + H]+ (C14H8O479Br281Br3, Δ 0 mmu). Compound 13: Rf 0.51 (Hex/EtOAc 8:1, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 2.24 (3H, s), 2.36 (3H, s), 7.01 (1H, d, J = 2.1 Hz), 7.63 (1H, d, J = 2.1 Hz), 8.13 (1H, s), 1H NMR (CDCl3) δ 2.25 (3H, s), 2.36 (3H, s), 6.58 (1H, d, J = 2.0 Hz), 7.47 (1H, d, J = 2.0 Hz), 7.87 (1H, s), HRESIMS m/z 704.6226 [M + Na]+ (C16H9O579Br281Br3Na, Δ 0.0023 mmu).
3.6. Debromination
HBr (10 mL) and Na2SO3 (20 equivalents) in AcOH (20 mL) were added to compound 1 (60 mg). The mixture was refluxed for 6 h and then neutralized with dilute KOH to pH 7 and partitioned using EtOAc and H2O. The organic layer formed was separated and purified by HPLC (RP18, MeOH) to give 1 (1.8 mg) and 2 (0.3 mg). Compound 1: Rf 0.51 (EtOAc/Hexane 1:2, n-silica, UV λ 254 nm); 1H NMR (Me2CO-d6) δ 6.83 (1H, d, J = 2.2 Hz), 7.40 (1H, d, J = 2.2 Hz). Compound 2: Rf 0.43 (EtOAc/Hexane 1:2, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 6.79 (1H, d, J = 2.3 Hz), 7.38 (1H, d, J = 2.3 Hz), 7.47 (1H, s).
HBr (2.5 mL) and Na2SO3 (15 equivalents) in AcOH (20 mL) were added to compound 1 (55.5 mg). The mixture was refluxed for 6 h and then dilute KOH was added to raise the pH to 10–11, after which it was partitioned using EtOAc and H2O. The organic layer formed was separated and purified by HPLC (RP18, MeOH, MeOH-H2O 2:1 + 0.1% TFA) to give 1 (1.26 mg), 2 (10.5 mg), 14 (1.2 mg), 16 (1.0 mg), 17 (2.8 mg), and 18 (0.5 mg). Compound 1: 1H NMR (CD3OD) δ 6.47 (1H, d, J = 2.2 Hz), 7.35 (1H, d, J = 2.2 Hz). Compound 14 in mixture form with 2. Compound 2: 1H NMR (CD3OD) δ 6.65 (1H, bs), 7.32 (1H, d, J = 2.3 Hz), 7.39 (1H, bs). Compound 14: 1H NMR (CD3OD) δ 6.45 (1H, d, J = 2.3 Hz), 7.31 (1H, d, J = 2.3 Hz), 7.38 (1H, s). Compound 16: 1H NMR (Me2CO-d6) δ 6.64 (1H, d, J = 2.5 Hz), 7.24 (1H, d, J = 2.5 Hz), 7.37 (1H, d, J = 2.5 Hz), 7.38 (1H, d, J = 2.5 Hz), HREIMS m/z 515.7007 [M]+ (C12H6O379Br381Br, Δ −0.0023 mmu). Compound 17: 1H NMR (Me2CO-d6) 6.69 (1H, d, J = 2.5 Hz), δ 6.74 (1H, d, J = 2.5 Hz), 7.27 (1H, d, J = 2.5 Hz,), 7.78 (1H, d, J = 2.5 Hz), LREIMS m/z 517.7 [M]+ (C12H6O381Br279Br2). Compound 18: 1H NMR (Me2CO-d6) δ 6.67 (d, overlapped), 7.03 (2H, dd, J = 8.5 Hz), 7.81 (1H, d, J = 2.5 Hz), LREIMS m/z 517.7 [M]+ (C12H6O379Br281Br2).
To 1 (60.6 mg) in AcOH HBr (10 mL) and Na2SO3 (20 equivalents) in AcOH (20 mL) were added. The mixture was refluxed for 24 h and then neutralized with dilute KOH to pH 7 and partitioned using EtOAc and water. The organic layer formed was separated and purified by HPLC (RP 18, MeCN + 0.1% TFA) to give 16 (4.1 mg) and 20 (0.5 mg). Compound 16: Rf 0.79 (EtOAc/Hexane 1:2, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 6.84 (1H, d, J = 2.3 Hz), 7.16 (1H, d, J = 2.3 Hz), 7.27 (1H, d, J = 2.3 Hz), 7.36 (1H, d, J = 2.3 Hz). Compound 20: Rf0.65 (EtOAc/Hexane 1:2, n-silica, UV λ 254 nm), 1H NMR (Me2CO-d6) δ 6.60 (1H, d, J = 2.3 Hz), 6.91 (1H, d, J = 8.5 Hz), 7.06 (1H, dd, J = 8.5, 2.3 Hz), 7.27 (1H, d, J = 2.3 Hz), 7.37 (1H, d, J = 2.3 Hz), HRESIMS m/z 441.2803 [M + H]+ C12H7O381Br3 (Δ −1.5159 mmu).
To compound 1 (60.1 mg) HBr (10 mL) and Na2SO3 (20 equivalents) in AcOH (20 mL) were added. The mixture was refluxed for 31 h and then the pH was raised to 10–11 with dilute KOH. The solution was partitioned using EtOAc and H2O. The organic layer was separated and purified by HPLC (RP18, MeOH, CH3CN-H2O 2:1) to give 17 (4.6 mg), 20 (2.2 mg), 23 (2.4 mg), and 26 (0.2 mg). Compound 20: 1H NMR (CD3OD) δ 6.74 (1H, d, J = 8.5 Hz), 6.79 (2H, q, J = 2.0 Hz), 6.97 (1H, dd, J = 2.0, 8.5 Hz), 7.07 (1H, d, J = 2.0 Hz). Compound 23: 1H NMR (Me2CO-d6) δ 6.67 (1H, d, J = 2.0 Hz), 6.75 (1H, d, J = 8.5 Hz), 6.93 (1H, dd, J = 2.0, 8.5 Hz), 7.03 (1H, d, J = 2.0 Hz), 7.12 (1H, d, J = 2.0 Hz). HREIMS m/z 435.7957 [M]+ C12H7O379Br281Br (Δ 0.0012 mmu). Compound 26: 1H NMR (CD3OD) δ 6.48 (1H, d, J = 8.0 Hz), 6.62 (1H, td, J = 8.0, 2.0 Hz), 6.87 (1H, dq, J = 8.0, 2.0 Hz), 6.87 (1H, d, J = 2.0 Hz), 7.05 (1H, d, J = 2.5 Hz), 7.22 (1H, d, J = 2.5 Hz). HREIMS m/z 357.8824 [M]+ C12H9O379Br2 (Δ −0.0016 mmu).
3.7. Computational Study
Equilibrium geometries were calculated for all structures using the ωB97X-D/6–311 + G(2d,p) density functional model implemented by Spartan ’20 (Wavefunction Inc, Irvine, CA, USA). Then, HOMO, LUMO, and electrostatic charge data can be used to support analysis of synthesized and natural compounds. Calculation of theoretical
13C NMR chemical shifts was performed according the protocol of Hehre et al. [
6], comprising six steps: (1) A conformer search using the MMFF molecular mechanic model was performed and high energy conformers (40 kJ/mol) were removed. (2) Equilibrium geometries were calculated using the HF/3-21G model and duplicate conformers were removed. Then, high-energy conformers (40 kJ/mol) were removed. (3) Energies were calculated using the ωB97X-D/6-31G* density functional model and high-energy conformers (15 kJ/mol) were removed. (4) Equilibrium geometries were calculated using the ωB97X-V/6–311 + G(2df,2p) (6–311G*) density functional model and high-energy conformers (10 kJ/mol) were removed. (5) Energies were calculated using the ωB97X-V/6–311 + G(2df,2p) (6–311G*) density functional model. (6)
13C NMR chemical shifts were calculated using the ωB97X-D/6–31G* followed by correction of
13C NMR chemical shifts based on the empirical parameters followed by correction
13C NMR chemical shifts based on the Boltzmann weight obtained in step 5.
3.8. X-ray Study
Single crystals of C
12H
6Br
6O
4 (
1), C
14H
8Br
6O
3 (
8), and C
14H
9Br
5O
3 (
9) were supplied. A suitable crystal was selected and mounted on Rigaku Saturn 724 Plus with AFC10. The crystal was kept at 113 K or 123.15 K during data collection. Using Olex2 [
31], the structure was solved with the SHELXT [
32] structure solution program using direct methods and refined with the SHELXL [
33] refinement package using least squares minimization.
Crystal data for 1, C12H6Br6O4 (M =693.63 g/mol): Monoclinic, space group P21/n (no. 14), a = 4.74979(10) Å, b = 18.7360(3) Å, c = 18.3463(4) Å, β = 91.6135(19)°, V = 1632.02(6) Å3, Z = 4, T = 123.15 K, μ(MoKα) = 14.772 mm−1, Dcalc = 2.823 g/cm3, 20780 reflections measured (3.108° ≤ 2Θ ≤62.354°), 4898 unique (Rint = 0.0374, Rsigma = 0.0252) which were used in all calculations. The final R1 was 0.0435 (I >2σ(I)) and wR2 was 0.1269 (all data).
Crystal data for 8, C14H8Br6O3 (M =703.66 g/mol): Triclinic, space group P-1 (no. 2), a = 8.733(3) Å, b = 8.911(3) Å, c = 12.769(4) Å, α = 104.185(4)°, β = 100.011(3)°, γ = 101.892(3)°, V = 916.0(5) Å3, Z = 2, T = 113 K, μ(MoKα) = 13.158 mm−1, Dcalc = 2.551 g/cm3, 7554 reflections measured (6.588° ≤ 2Θ ≤54.968°), 4046 unique (Rint = 0.0369, Rsigma = 0.0427) which were used in all calculations. The final R1 was 0.0422 (I >2σ(I)) and wR2 was 0.0997 (all data).
Crystal data for 9, C14H9Br5O3 (M =624.76 g/mol): Orthorhombic, space group Pca21 (no. 29), a = 6.9014(15) Å, b = 11.940(3) Å, c = 21.090(5) Å, V = 1737.9(7) Å3, Z = 4, T = 113.15 K, μ(MoKα) = 11.569 mm−1, Dcalc = 2.388 g/cm3, 17770 reflections measured (6.82° ≤ 2Θ ≤ 54.952°), 3584 unique (Rint = 0.0623, Rsigma = 0.0509) which were used in all calculations. The final R1 was 0.0359 (I > 2σ(I)) and wR2 was 0.0681 (all data).
3.9. Agar-Plate Diffusion Assay
Staphylococcus aureus ATCC 6538 and
Klebsiella pneumoniae were used for biological evaluation. Concentrations assayed ranged from 0.08 to 1.25 µg/disks [
14,
15]. DMSO was used to dissolve the compounds, while vancomycin and oxacillin were used as positive controls for
Staphylococcus aureus and gentamycin was used as a positive control for
Klebsiella pneumoniae. The minimum inhibitory dose (MID, μg/disk) was defined as the minimum dose that induced an obvious inhibition zone (1–1.5 mm) [
28]. The disk diameter was 6 mm.
3.10. In Vitro Cytotoxicity Assay
In vitro cytotoxicity was determined against human embryonic kidney (HEK293T) cells. The assay was performed in 96-well treated tissue culture plates. Cells were seeded in the wells (5000 cells in 100 µL media containing RPMI1640, FBS, penicillin and streptomycin) and incubated for 24 h. Samples were then added and plates were again incubated for 48 h. MTT was then added and the plates were incubated for 4 h at 37 °C. Formazan crystals formed were dissolved in EtOH and absorbances were read at λ = 595 nm. The result was analyzed by using Prism 9 software (Graphpad, San Diego, CA, USA) to obtain IC50 values.


 ,
, 













{kind=link}
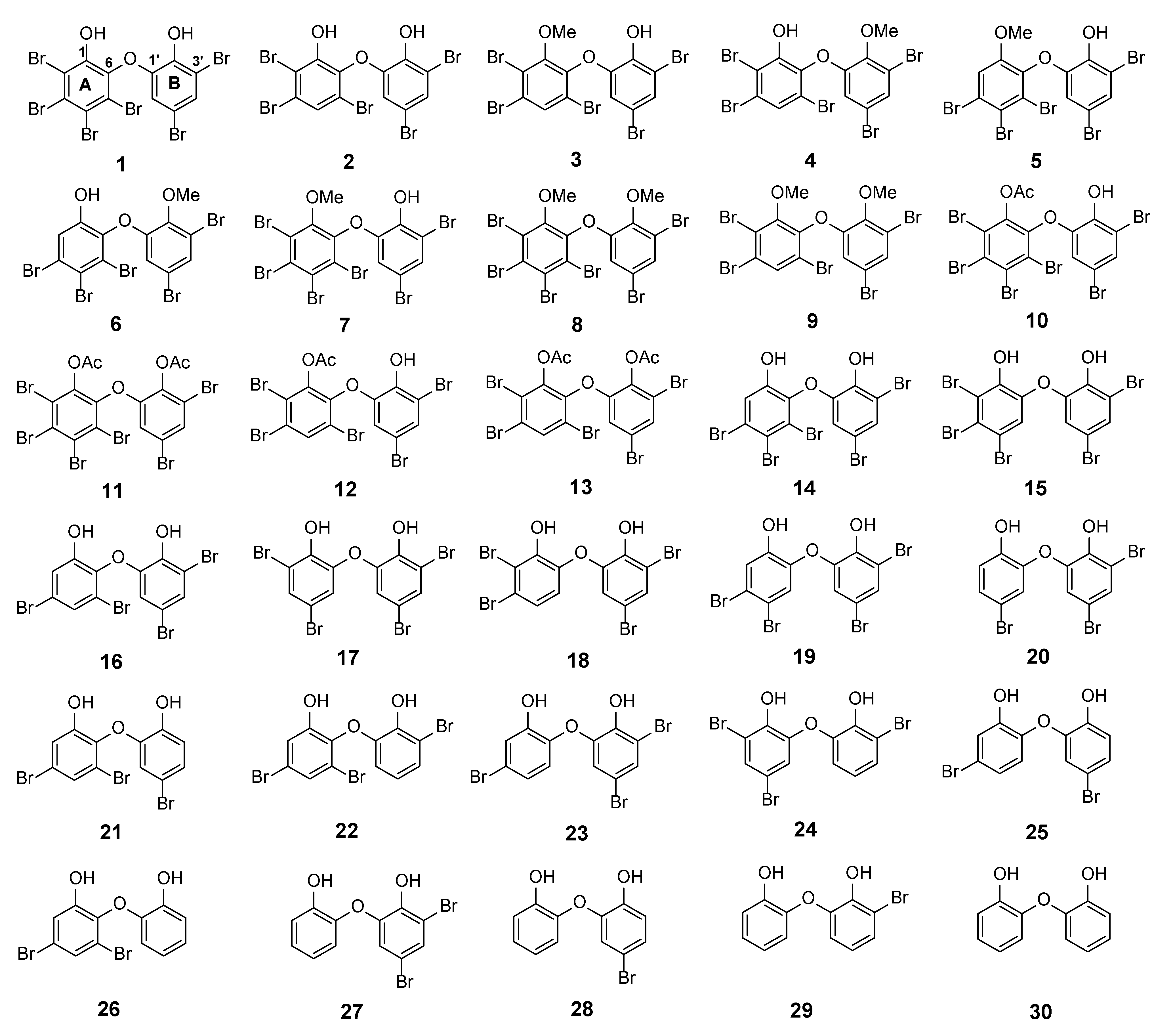{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}