
Isothiourea-Catalyzed Enantioselective α-Alkylation of Esters via 1,6-Conjugate Addition to para-Quinone Methides

 and
and
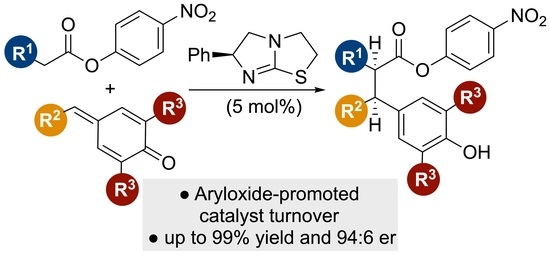
Abstract
:
1. Introduction
2. Results
2.1. Reaction Optimization
2.2. Reaction Scope and Limitations
2.3. Proposed Mechanism
3. Materials and Methods
3.1. General Procedure for the Enantioselective 1,6-Addition
3.2. Representative Synthesis and Characterization of Compounds 7 and 8 (Entry 8)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References and Notes
- Rokita, S.E. Quinone Methides; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Toteva, M.M.; Richard, J.P. The generation and reactions of quinone methides. Adv. Phys. Org. Chem. 2011, 45, 39–91. [Google Scholar] [PubMed] [Green Version]
- Turner, A.B. Quinone methides. Q. Rev. Chem. Soc. 1964, 18, 347–360. [Google Scholar] [CrossRef]
- Wagner, H.-U.R. Gompper. In The Chemistry of Quinonoid Compounds; Wiley: London, UK, 1974; pp. 1145–1178. [Google Scholar]
- Richter, D.; Hampel, N.; Singer, T.; Ofial, A.R.; Mayr, H. Synthesis and Characterization of Novel Quinone Methides: Reference Electrophiles for the Construction of Nucleophilicity Scales. Eur. J. Org. Chem. 2009, 3203–3211. [Google Scholar] [CrossRef]
- Singh, M.S. Reactive Intermediates in Organic Chemistry: Structure and Mechanism; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Freccero, M. Quinone methides as alkylating and cross-linking agents. Mini Rev. Org. Chem. 2004, 1, 403–415. [Google Scholar] [CrossRef]
- Peters, M.G. Chemical modifications of biopolymers by quinones and quinone methides. Angew. Chem. Int. Ed. 1989, 28, 555–570, Angew. Chem.1989, 101, 572–587. [Google Scholar] [CrossRef]
- Martin, H.J.; Magauer, T.; Mulzer, J. In Pursuit of a Competitive Target: Total Synthesis of the Antibiotic Kendomycin. Angew. Chem. Int. Ed. 2010, 49, 5614–5626, Angew. Chem.2010, 122, 5746–5758. [Google Scholar] [CrossRef]
- Jansen, R.; Gerth, K.; Steinmetz, H.; Reinecke, S.; Kessler, W.; Kirschning, A.; Müller, R. Elansolid A3, a Unique p-Quinone Methide Antibiotic from Chitinophaga sancti. Chem. Eur. J. 2011, 17, 7739–7744. [Google Scholar] [CrossRef]
- Dehn, R.; Katsuyama, Y.; Weber, A.; Gerth, K.; Jansen, R.; Steinmetz, H.; Höfle, G.; Müller, R.; Kirschning, A. Molecular Basis of Elansolid Biosynthesis: Evidence for an Unprecedented Quinone Methide Initiated Intramolecular Diels–Alder Cycloaddition/Macrolactonization. Angew. Chem. Int. Ed. 2011, 50, 3882–3887, Angew. Chem.2011, 123, 3968–3973. [Google Scholar] [CrossRef]
- Gai, K.; Fang, X.; Li, X.; Xu, J.; Wu, X.; Lin, A.; Yao, H. Synthesis of spiro[2.5]octa-4,7-dien-6-one with consecutive quaternary centers via 1,6-conjugate addition induced dearomatization of para-quinone methides. Chem. Commun. 2015, 51, 15831–15834. [Google Scholar] [CrossRef]
- Lόpez, A.; Parra, A.; Jarava-Barrera, C.; Tortosa, M. Copper-catalyzed silylation of p-quinone methides: New entry to dibenzylic silanes. Chem. Commun. 2015, 51, 17684–17687. [Google Scholar] [CrossRef] [Green Version]
- Ramanjaneyulu, B.T.; Mahesh, S.; Anand, R.V. Bis(amino)cyclopropenylidene-Catalyzed 1,6-Conjugate Addition of Aromatic Aldehydes to para-Quinone Methides: Expedient Access to α,α′-Diarylated Ketones. Org. Lett. 2015, 17, 3952–3955. [Google Scholar] [CrossRef]
- Yuan, Z.; Fang, X.; Li, X.; Wu, J.; Yao, H.; Lin, A. 1,6-Conjugated Addition-Mediated [2+1] Annulation: Approach to Spiro[2.5]octa-4,7-dien-6-one. J. Org. Chem. 2015, 80, 11123–11130. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.; Anand, R.V. Expedient Access to Unsymmetrical Diarylindolylmethanes through Palladium-Catalyzed Domino Electrophilic Cyclization–Extended Conjugate Addition Approach. Org. Lett. 2015, 17, 3390–3393. [Google Scholar] [CrossRef]
- Roiser, L.; Zielke, K.; Waser, M. Enantioselective Spirocyclopropanation of para-Quinone Methides using Ammonium Ylides. Org. Lett. 2017, 19, 2338–2341. [Google Scholar] [CrossRef]
- Roiser, L.; Zielke, K.; Waser, M. Formal (4+1) Cyclization of Ammonium Ylides with Vinylogous para-Quinone Methides. Synthesis 2018, 50, 4047–4054. [Google Scholar]
- Caruana, L.; Fochi, M.; Bernardi, L. The Emergence of Quinone Methides in Asymmetric Organocatalysis. Molecules 2015, 20, 11733–11764. [Google Scholar] [CrossRef]
- Parra, A.; Tortosa, M. para-Quinone Methide: A New Player in Asymmetric Catalysis. ChemCatChem 2015, 7, 1524–1526. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, P.; Kaya, U.; Enders, D. Advances in Organocatalytic 1,6-Addition Reactions: Enantioselective Construction of Remote Stereogenic Centers. Adv. Synth. Catal. 2017, 359, 888–912. [Google Scholar] [CrossRef]
- Li, W.; Xu, X.; Zhang, P.; Li, P. Recent Advances in the Catalytic Enantioselective Reactions of para-Quinone Methides. Chem. Asian J. 2018, 13, 2350–2359. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Hao, W.-J.; Tu, S.-J.; Jiang, B. Recent developments in 1,6-addition reactions of para-quinone methides (p-QMs). Org. Chem. Front. 2020, 7, 1743–1778. [Google Scholar] [CrossRef]
- Wang, Z.; Wong, Y.F.; Sun, J. Catalytic Asymmetric 1,6-Conjugate Addition of para-Quinone Methides: Formation of All-Carbon Quaternary Stereocenters. Angew. Chem. Int. Ed. 2015, 54, 13711–13714, Angew. Chem.2015, 127, 13915–13918. [Google Scholar] [CrossRef]
- Dong, N.; Zhang, Z.-P.; Xue, X.-S.; Li, X.; Cheng, J.-P. Phosphoric Acid Catalyzed Asymmetric 1,6-Conjugate Addition of Thioacetic Acid to para-Quinone Methides. Angew. Chem. Int. Ed. 2016, 55, 1460–1464, Angew. Chem.2016, 128, 1482–1486. [Google Scholar] [CrossRef]
- Wong, Y.F.; Wang, Z.; Sun, J. Chiral phosphoric acid catalyzed asymmetric addition of naphthols to para-quinone methides. Org. Biomol. Chem. 2016, 14, 5751–5754. [Google Scholar] [CrossRef]
- Chen, M.; Sun, J. How understanding the role of an additive can lead to an improved synthetic protocol without an additive: Organocatalytic synthesis of chiral diarylmethyl alkynes. Angew. Chem. 2017, 129, 12128–12132, Angew. Chem. Int. Ed.2017, 56, 11966–11970. [Google Scholar] [CrossRef]
- Yan, J.; Chen, M.; Sung, H.H.-Y.; Williams, I.D.; Sun, J. An Organocatalytic Asymmetric Synthesis of Chiral β,β-Diaryl-α-amino Acids via Addition of Azlactones to In Situ Generated para-Quinone Methides. Chem. Asian J. 2018, 13, 2440–2444. [Google Scholar] [CrossRef]
- Rahman, A.; Zhou, Q.; Lin, X. Asymmetric organocatalytic synthesis of chiral 3,3-disubstituted oxindoles via a 1, 6-conjugate addition reaction. Org. Biomol. Chem. 2018, 16, 5301–5309. [Google Scholar] [CrossRef]
- Li, W.; Xu, X.; Liu, Y.; Gao, H.; Cheng, Y.; Li, P. Enantioselective Organocatalytic 1,6-Addition of Azlactones to para-Quinone Methides: An Access to α,α-Disubstituted and β,β-Diaryl-α-amino acid Esters. Org. Lett. 2018, 20, 1142–1145. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-R.; Jiang, X.-L.; Hang, Q.-Q.; Zhang, S.; Mei, G.-J.; Shi, F. Catalytic Asymmetric Conjugate Addition of Indoles to para-Quinone Methide Derivatives. J. Org. Chem. 2019, 84, 7829–7839. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, Y.; Pan, X.; Wang, G.; Liu, L. Synthesis of Chiral Triarylmethanes Bearing All-Carbon Quaternary Stereocenters: Catalytic Asymmetric Oxidative Cross-Coupling of 2,2-Diarylacetonitriles and (Hetero)arenes. Angew. Chem. 2020, 132, 3077–3081, Angew. Chem.2020, 59, 3053–3057. [Google Scholar] [CrossRef]
- Niu, J.-P.; Nie, J.; Li, S.; Ma, J.-A. Organocatalytic asymmetric synthesis of β,β-diaryl ketones via one-pot tandem dehydration/1,6-addition/decarboxylation transformation of β-keto acids and 4-hydroxybenzyl alcohols. Chem. Commun. 2020, 56, 8687–8690. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhi, Y.; Wang, A.; Enders, D. Asymmetric Organocatalytic Synthesis of 3-Diarylmethine-Substituted Oxindoles Bearing a Quaternary Stereocenter via 1,6-Conjugate Addition to para-Quinone Methides. ACS Catal. 2016, 6, 657–660. [Google Scholar] [CrossRef]
- Li, X.; Xu, X.; Wei, W.; Lin, A.; Yao, H. Organocatalyzed Asymmetric 1,6-Conjugate Addition of para-Quinone Methides with Dicyanoolefins. Org. Lett. 2016, 18, 428–431. [Google Scholar] [CrossRef]
- Deng, Y.-H.; Zhang, X.-Z.; Yu, K.-Y.; Yan, X.; Du, J.-Y.; Huang, H.; Fan, C.-A. Bifunctional tertiary amine-squaramide catalyzed asymmetric catalytic 1,6-conjugate addition/aromatization of para-quinone methides with oxindoles. Chem. Commun. 2016, 52, 4183–4186. [Google Scholar] [CrossRef] [PubMed]
- Toràn, R.; Vila, C.; Sanz-Marco, A.; Muñoz, M.C.; Pedro, J.R.; Blay, G. Organocatalytic Enantioselective 1,6-aza-Michael Addition of Isoxazolin-5-ones to p-Quinone Methides. Eur. J. Org. Chem. 2020, 2020, 627–630. [Google Scholar] [CrossRef]
- Wang, L.; Yang, F.; Xua, X.; Jiang, J. Organocatalytic 1,6-hydrophosphination of para-quinone methides: Enantioselective access to chiral 3-phosphoxindoles bearing phosphorus-substituted quaternary carbon stereocenters. Org. Chem. Front. 2021, 8, 2002–2008. [Google Scholar] [CrossRef]
- Caruana, L.; Kniep, F.; Johansen, T.K.; Poulsen, H.P.; Jørgensen, K.A. A New Organocatalytic Concept for Asymmetric α-Alkylation of Aldehydes. J. Am. Chem. Soc. 2014, 136, 15929–15932. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Huang, B.; Zhou, T.; Tao, H.; Xiao, Y.; Liu, L.; Zhang, J. Phosphine-Catalyzed Asymmetric Intermolecular Cross-Vinylogous Rauhut–Currier Reactions of Vinyl Ketones with para-Quinone Methides. ACS Catal. 2017, 7, 2805–2809. [Google Scholar] [CrossRef]
- Kang, T.-C.; Wu, L.-P.; Yu, Q.-W.; Wu, X.-Y. Enantioselective Rauhut–Currier-Type 1,6-Conjugate Addition of Methyl Vinyl Ketone to para-Quinone Methides. Chem. Eur. J. 2017, 23, 6509–6513. [Google Scholar] [CrossRef]
- Wang, D.; Song, Z.-F.; Wang, W.-J.; Xu, T. Highly Regio- and Enantioselective Dienylation of p-Quinone Methides Enabled by an Organocatalyzed Isomerization/Addition Cascade of Allenoates. Org. Lett. 2019, 21, 3963–3967. [Google Scholar] [CrossRef]
- Li, W.; Yuan, H.; Liu, Z.; Zhang, Z.; Cheng, Y.; Li, P. NHC-Catalyzed Enantioselective [4+3] Cycloaddition of Ortho-Hydroxyphenyl Substituted Para-Quinone Methides with Isatin-Derived Enals. Adv. Synth. Catal. 2018, 360, 2460–2464. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, X.-Y.; Rissanen, K.; Ender, D. Asymmetric synthesis of spiro-oxindole-ε-lactones through N-heterocyclic carbene catalysis. Org. Lett. 2018, 20, 3622–3626. [Google Scholar] [CrossRef]
- Zhao, M.-X.; Xiang, J.; Zhao, Z.-Q.; Zhao, X.-L.; Shi, M. Asymmetric synthesis of dihydrocoumarins via catalytic sequential 1,6-addition/transesterification of α-isocyanoacetates with para-quinone methides. Org. Biomol. Chem. 2020, 18, 1637–1646. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Pradhan, S.; Kumar, K.; Chatterjee, I. Asymmetric organocatalytic double 1,6-addition: Rapid access to chiral chromans with molecular complexity. Org. Chem. Front. 2020, 7, 1388–1394. [Google Scholar] [CrossRef]
- Chu, W.D.; Zhang, L.F.; Bao, X.; Zhao, X.H.; Zeng, C.; Du, J.Y.; Zhang, G.B.; Wang, F.X.; Ma, X.Y.; Fan, C.A. Asymmetric Catalytic 1,6-Conjugate Addition/Aromatization of para-Quinone Methides: Enantioselective Introduction of Functionalized Diarylmethine Stereogenic Centers. Angew. Chem. Int. Ed. 2013, 52, 9229–9233, Angew. Chem.2013, 125, 9399–9403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Z.; Deng, Y.-H.; Yan, X.; Yu, K.-Y.; Wang, F.-X.; Ma, X.-Y.; Fan, C.-A. Diastereoselective and Enantioselective Synthesis of Unsymmetric β,β-Diaryl-α-Amino Acid Esters via Organocatalytic 1,6-Conjugate Addition of para-Quinone Methides. J. Org. Chem. 2016, 81, 5655–5662. [Google Scholar] [CrossRef]
- Ge, L.; Lu, X.; Cheng, C.; Chen, J.; Cao, W.; Wu, X.; Zhao, G. Amide-Phosphonium Salt as Bifunctional Phase Transfer Catalyst for Asymmetric 1,6-Addition of Malonate Esters to para-Quinone Methides. J. Org. Chem. 2016, 81, 9315–9325. [Google Scholar] [CrossRef]
- Santra, S.; Porey, A.; Jana, B.; Guin, J. N-Heterocyclic carbenes as chiral Brønsted base catalysts: A highly diastereo- and enantioselective 1,6-addition reaction. Chem. Sci. 2018, 9, 6446–6450. [Google Scholar] [CrossRef] [Green Version]
- Eitzinger, A.; Winter, M.; Schörgenhumer, J.; Waser, M. Quaternary β 2, 2-amino acid derivatives by asymmetric addition of isoxazolidin-5-ones to para-quinone methides. Chem. Commun. 2020, 56, 579–582. [Google Scholar] [CrossRef] [Green Version]
- Van, K.N.; Morrill, L.C.; Smith, A.D.; Romo, D. Lewis Base Catalysis in Organic Synthesis; Vedejs, E., Denmark, S.E., Eds.; Wiley-VCH: Weinheim, Germany, 2016; Chapter 13; pp. 527–653. [Google Scholar]
- Morrill, L.C.; Smith, A.D. Organocatalytic Lewis base functionalisation of carboxylic acids, esters and anhydrides via C1-ammonium or azolium enolates. Chem. Soc. Rev. 2014, 43, 6214–6226. [Google Scholar] [CrossRef] [Green Version]
- Gaunt, M.J.; Johansson, C.C.C. Recent developments in the use of catalytic asymmetric ammonium enolates in chemical synthesis. Chem. Rev. 2007, 107, 5596–5605. [Google Scholar] [CrossRef]
- McLaughlin, C.; Smith, A.D. Generation and Reactivity of C (1)-Ammonium Enolates by Using Isothiourea Catalysis. Chem. Eur. J. 2021, 27, 1533–1555. [Google Scholar] [CrossRef] [PubMed]
- Young, C.M.; Stark, D.G.; West, T.H.; Taylor, J.E.; Smith, A.D. Exploiting the Imidazolium Effect in Base-free Ammonium Enolate Generation: Synthetic and Mechanistic Studies. Angew. Chem. 2016, 128, 14606–14611, Angew. Chem.2016, 55, 14394–14399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.; Chen, X.; Chen, S.; Jiang, K.; Torres, J.; Chi, Y.R. Access to pyridines via DMAP-catalyzed activation of α-chloro acetic ester to react with unsaturated imines. Org. Chem. Front. 2014, 1, 148–150. [Google Scholar] [CrossRef]
- Young, C.M.; Taylor, J.E.; Smith, A.D. Evaluating aryl esters as bench-stable C (1)-ammonium enolate precursors in catalytic, enantioselective Michael addition–lactonisations. Org. Biomol. Chem. 2019, 17, 4747–4752. [Google Scholar] [CrossRef] [PubMed]
- Hartley, W.C.; O’Riordan, T.J.C.; Smith, A.D. Aryloxide-Promoted Catalyst Turnover in Lewis Base Organocatalysis. Synthesis 2017, 49, 3303–3310. [Google Scholar]
- West, T.H.; Daniels, D.S.B.; Slawin, A.M.Z.; Smith, A.D. An isothiourea-catalyzed asymmetric [2, 3]-rearrangement of allylic ammonium ylides. J. Am. Chem. Soc. 2014, 136, 4476–4479. [Google Scholar] [CrossRef] [Green Version]
- West, T.H.; Walden, D.M.; Taylor, J.E.; Brueckner, A.C.; Johnston, R.C.; Cheong, P.H.-Y.; Lloyd-Jones, G.C.; Smith, A.D. Catalytic Enantioselective [2, 3]-Rearrangements of Allylic Ammonium Ylides: A Mechanistic and Computational Study. J. Am. Chem. Soc. 2017, 139, 4366–4375. [Google Scholar] [CrossRef] [Green Version]
- Spoehrle, S.S.M.; West, T.H.; Taylor, J.E.; Slawin, A.M.Z.; Smith, A.D. Tandem Palladium and Isothiourea Relay Catalysis: Enantioselective Synthesis of α-Amino Acid Derivatives via Allylic Amination and [2, 3]-Sigmatropic Rearrangement. J. Am. Chem. Soc. 2017, 139, 11895–11902. [Google Scholar] [CrossRef] [Green Version]
- Kasten, K.; Slawin, A.M.Z.; Smith, A.D. Enantioselective Synthesis of β-Fluoro-β-aryl-α-aminopentenamides by Organocatalytic [2, 3]-Sigmatropic Rearrangement. Org. Lett. 2017, 19, 5182–5185. [Google Scholar] [CrossRef] [Green Version]
- West, T.H.; Spoehrle, S.S.M.; Smith, A.D. Isothiourea-catalysed chemo-and enantioselective [2, 3]-sigmatropic rearrangements of N, N-diallyl allylic ammonium ylides. Tetrahedron 2017, 73, 4138–4149. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.J.; Amos, J.L.; Klein, J.C.; Do, D.T.; Snaddon, T.N. Uniting C1-ammonium enolates and transition metal electrophiles via cooperative catalysis: The direct asymmetric α-allylation of aryl acetic acid esters. J. Am. Chem. Soc. 2016, 138, 5214–5217. [Google Scholar] [CrossRef]
- Schwarz, K.J.; Pearson, C.M.; Cintron-Rosado, G.A.; Liu, P.; Snaddon, T.N. Traversing Steric Limitations by Cooperative Lewis Base/Palladium Catalysis: An Enantioselective Synthesis of α-Branched Esters Using 2-Substituted Allyl Electrophiles. Angew. Chem. 2018, 130, 7926–7929, Angew. Chem.2018, 57, 7800–7803. [Google Scholar] [CrossRef]
- Scaggs, W.R.; Snaddon, T.N. Enantioselective α-Allylation of Acyclic Esters using B(pin)-Substituted Electrophiles: Independent Regulation of Stereocontrol Elements via Cooperative Pd/Lewis Base Catalysis. Chem. Eur. J. 2018, 24, 14378–14381. [Google Scholar] [CrossRef]
- Fyfe, J.W.B.; Kabia, O.M.; Pearson, C.M.; Snaddon, T.N. Si-directed regiocontrol in asymmetric Pd-catalyzed allylic alkylations using C1-ammonium enolate nucleophiles. Tetrahedron 2018, 74, 5383–5391. [Google Scholar] [CrossRef]
- Hutchings-Goetz, J.; Yang, C.; Snaddon, T.N. Enantioselective Syntheses of Strychnos and Chelidonium Alkaloids through Regio- and Stereocontrolled Cooperative Catalysis. ACS Catal. 2018, 8, 10537–10544. [Google Scholar] [CrossRef] [Green Version]
- Pearson, C.M.; Fyfe, J.W.B.; Snaddon, T.N. A Regio- and Stereodivergent Synthesis of Homoallylic Amines by a One-Pot Cooperative-Catalysis-Based Allylic Alkylation/Hofmann Rearrangement Strategy. Angew. Chem. Int. Ed. 2019, 58, 10521–10527, Angew. Chem.2019, 131, 10631–10637. [Google Scholar] [CrossRef]
- Scaggs, W.R.; Scaggs, T.D.; Snaddon, T.N. An enantioselective synthesis of α-alkylated pyrroles via cooperative isothiourea/palladium catalysis. Org. Biomol. Chem. 2019, 17, 1787–1790. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.J.; Yang, C.; Fyfe, J.W.B.; Snaddon, T.N. Enantioselective α-Benzylation of Acyclic Esters Using π-Extended Electrophiles. Angew. Chem. Int. Ed. 2018, 57, 12102–12105, Angew. Chem.2018, 130, 12278–12281. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Beiger, J.J.; Hartwig, J.F. Stereodivergent allylic substitutions with aryl acetic acid esters by synergistic iridium and Lewis base catalysis. J. Am. Chem. Soc. 2017, 139, 87–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Zhang, Z.J.; Gong, L.Z. Asymmetric [4+2] Annulation of C1 Ammonium Enolates with Copper-Allenylidenes. Angew. Chem. Int. Ed. 2017, 56, 5212–5216, Angew. Chem.2017, 129, 5296–5300. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, Z.J.; Chen, S.S.; Fan, T.; Gong, L.Z. Lewis base/copper cooperatively catalyzed asymmetric α-amination of esters with diaziridinone. J. Am. Chem. Soc. 2018, 140, 3177–3180. [Google Scholar] [CrossRef] [PubMed]
- Arokianathar, J.N.; Frost, A.B.; Slawin, A.M.Z.; Stead, D.; Smith, A.D. Isothiourea-Catalyzed Enantioselective Addition of 4-Nitrophenyl Esters to Iminium Ions. ACS Catal. 2018, 8, 1067–1075. [Google Scholar] [CrossRef]
- Zhao, F.; Shu, C.; Young, C.M.; Carpenter-Warren, C.; Slawin, A.M.Z.; Smith, A.D. Enantioselective Synthesis of α-Aryl-β2-Amino-Esters by Cooperative Isothiourea and Brønsted Acid Catalysis. Angew. Chem. Int. Ed. 2021, 60, 11892–11900, Angew. Chem.2021, 133, 11999–12007. [Google Scholar] [CrossRef]
- McLaughlin, C.; Slawin, A.M.Z.; Smith, A.D. Base-free Enantioselective C (1)-Ammonium Enolate Catalysis Exploiting Aryloxides: A Synthetic and Mechanistic Study. Angew. Chem. Int. Ed. 2019, 131, 15255–15263, Angew. Chem.2019, 58, 15111–15119. [Google Scholar] [CrossRef]
- McLaughlin, C.; Bitai, J.; Barber, L.; Slawin, A.M.Z.; Smith, A.D. Catalytic enantioselective synthesis of 1, 4-dihydropyridines via the addition of C (1)-ammonium enolates to pyridinium salts. Chem. Sci. 2021. accepted for publication. [Google Scholar] [CrossRef]
- Stockhammer, L.; Weinzierl, D.; Bögl, T.; Waser, M. Enantioselective α-Chlorination Reactions of in Situ Generated C1 Ammonium Enolates under Base-Free Conditions. Org. Lett. 2021, 23, 6143–6147. [Google Scholar] [CrossRef]
- Kim, B.; Kim, Y.; Lee, S.Y. Stereodivergent Carbon–Carbon Bond Formation between Iminium and Enolate Intermediates by Synergistic Organocatalysis. J. Am. Chem. Soc. 2021, 143, 73–79. [Google Scholar]
- Yuan, S.; Liao, C.; Zheng, W.-H. Paracyclophane-Based Isothiourea-Catalyzed Highly Enantioselective α-Fluorination of Carboxylic Acids. Org. Lett. 2021, 23, 4142–4146. [Google Scholar] [CrossRef]
- Crystallographic data for (2S,1′R)-19 (CCDC 1992504), Cambridge Crystallographic Data Centre.
- The relative and absolute configuration of all other products was assigned by analogy, with consistent diagnostic 1H NMR signals of the proton at C(2) and C(1ʹ) stereocentres, indicating the same major diastereoisomer in each case.
- Morrill, L.C.; Douglas, J.; Lebl, T.; Slawin, A.M.Z.; Fox, D.J.; Smith, A.D. Isothiourea-mediated asymmetric Michael-lactonisation of trifluoromethylenones: A synthetic and mechanistic study. Chem. Sci. 2013, 4, 4146–4155. [Google Scholar] [CrossRef]
- Matviistsuk, A.; Greenhalgh, M.D.; Antúnez, D.-J.B.; Slawin, A.M.Z.; Smith, A.D. Aryloxide-Facilitated Catalyst Turnover in Enantioselective α, β-Unsaturated Acyl Ammonium Catalysis. Angew. Chem. Int. Ed. 2017, 56, 12282–12287, Angew. Chem.2017, 129, 12450–12455. [Google Scholar] [CrossRef]
- Catalyst turnover could also be envisaged through intramolecular nucleophilic displacement by the pendant phenol to give a cyclobutanone intermediate via a formal [2+2]-cycloaddition, which may then be ring-opened by para-nitrophenoxide to give the final product. Based on the high steric congestion of this proposed cyclobutanone intermediate, the mechanism given in Scheme 5 is considered more probable.
- Liu, P.; Yang, X.; Birman, V.B.; Houk, K.N. Origin of enantioselectivity in benzotetramisole-catalyzed dynamic kinetic resolution of azlactones. Org. Lett. 2012, 14, 3288–3291. [Google Scholar] [CrossRef] [PubMed]
- Abbasov, M.E.; Hudson, B.M.; Tantillo, D.J.; Romo, D. Acylammonium salts as dienophiles in Diels–Alder/lactonization organocascades. J. Am. Chem. Soc. 2014, 136, 4492–4495. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.R.T.; Walden, D.M.; Fallan, C.; Greenhalgh, M.D.; Cheong, P.H.-Y.; Smith, A.D. Non-bonding 1,5-S⋯O interactions govern chemo- and enantioselectivity in isothiourea-catalyzed annulations of benzazoles. Chem. Sci. 2016, 7, 6919–6927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenhalgh, M.D.; Smith, S.M.; Walden, D.M.; Taylor, J.E.; Brice, Z.; Robinson, E.R.T.; Fallan, C.; Cordes, D.B.; Slawin, A.M.Z.; Richardson, H.C.; et al. AC= O⋅⋅⋅ Isothiouronium Interaction Dictates Enantiodiscrimination in Acylative Kinetic Resolutions of Tertiary Heterocyclic Alcohols. Angew. Chem. Int. Ed. 2018, 57, 3200–3206, Angew. Chem.2018, 130, 3254–3260. [Google Scholar] [CrossRef] [Green Version]
- Young, C.M.; Elmi, A.; Pascoe, D.J.; Morris, R.K.; McLaughlin, C.; Woods, A.M.; Frost, A.B.; de la Houpliere, A.; Ling, K.B.; Smith, T.K.; et al. The Importance of 1, 5-Oxygen Chalcogen Interactions in Enantioselective Isochalcogenourea Catalysis. Angew. Chem. 2020, 132, 3734–3739, Angew. Chem.2020, 59, 3705–3710. [Google Scholar] [CrossRef]
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The origin of chalcogen-bonding interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef] [Green Version]
- Nagao, Y.; Miyamoto, S.; Miyamoto, M.; Takeshige, H.; Hayashi, K.; Sano, S.; Shiro, M.; Yamaguchi, K.; Sei, Y. Highly Stereoselective Asymmetric Pummerer Reactions That Incorporate Intermolecular and Intramolecular Nonbonded S⋯O Interactions. J. Am. Chem. Soc. 2006, 128, 9722–9729. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef]
- Breugst, M.; Koenig, J.J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 34, 5473–5487. [Google Scholar] [CrossRef]
- Bleiholder, C.; Gleiter, R.; Werz, D.B.; Köppel, H. Theoretical Investigations on Heteronuclear Chalcogen—Chalcogen Interactions: On the Nature of Weak Bonds between Chalcogen Centers. Inorg. Chem. 2007, 46, 2249–2260. [Google Scholar] [CrossRef]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F. From noncovalent chalcogen–chalcogen interactions to supramolecular aggregates: Experiments and calculations. Bleiholder. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef] [PubMed]
- Benz, S.; López-Andarias, J.; Mareda, J.; Sakai, N.; Matile, S. Catalysis with chalcogen bonds. Angew. Chem. 2017, 129, 830–833, Angew. Chem.2017, 56, 812–815. [Google Scholar] [CrossRef] [Green Version]
- Wonner, P.; Vogel, L.; Düser, M.; Gomes, L.; Kniep, F.; Mallick, B.; Werz, D.B.; Huber, S.M. Carbon–Halogen Bond Activation by Selenium-Based Chalcogen Bonding. Angew. Chem. Int. Ed. 2017, 56, 12009–12012, Angew. Chem.2017, 129, 12172–12176. [Google Scholar] [CrossRef] [PubMed]
- Wonner, P.; Vogel, L.; Kniep, F.; Huber, S.M. Catalytic Carbon–Chlorine Bond Activation by Selenium-Based Chalcogen Bond Donors. Chem. Eur. J. 2017, 23, 16972–16975. [Google Scholar] [CrossRef]
- Wonner, P.; Dreger, A.; Vogel, L.; Engelage, E.; Huber, S.M. Chalcogen Bonding Catalysis of a Nitro-Michael Reaction. Angew. Chem. Int. Ed. 2019, 58, 16923–16927, Angew. Chem.2019, 131, 17079–17083. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, H.; Liu, S.; Zhao, Z.; Zhang, L.; Hao, J.; Wang, Y. Chalcogen–chalcogen bonding catalysis enables assembly of discrete molecules. J. Am. Chem. Soc. 2019, 141, 9175–9179. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhu, H.; Feng, L.; Yu, Q.; Hao, J.; Zhu, R.; Wang, Y. Dual Chalcogen–Chalcogen Bonding Catalysis. J. Am. Chem. Soc. 2020, 142, 3117–3124. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | 6 (mol%) | Aryl Ester | Base | Yield (%) | dr (7:8) | er (7) | er (8) |
|---|---|---|---|---|---|---|---|
| 1 | 20 | 1 | i-Pr2NEt | 82 | 55:45 | 97:3 | 94:6 |
| 2 | 0 | 1 | i-Pr2NEt | 0 | - | - | - |
| 3 | 20 | 1 | Et3N | 98 | 60:40 | 97:3 | 94:6 |
| 4 | 20 | 2 | Et3N | 99 | 65:35 | 81:19 | 94:6 |
| 5 | 20 | 3 | Et3N | 88 | 60:40 | 94:6 | 91:9 |
| 6 | 20 | 4 | Et3N | 31 | 55:45 | 84:16 | 71:29 |
| 7 | 5 | 1 | Et3N | 95 | 55:45 | 93:7 | 88:12 |
| 8 [a] | 5 | 1 | Et3N | 99 | 60:40 | 92:8 | 85:15 |
| 9 [a] | 5 | 1 | none | 90 | 60:40 | 93:7 | 89:11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arokianathar, J.N.; Hartley, W.C.; McLaughlin, C.; Greenhalgh, M.D.; Stead, D.; Ng, S.; Slawin, A.M.Z.; Smith, A.D. Isothiourea-Catalyzed Enantioselective α-Alkylation of Esters via 1,6-Conjugate Addition to para-Quinone Methides. Molecules 2021, 26, 6333. https://doi.org/10.3390/molecules26216333
Arokianathar JN, Hartley WC, McLaughlin C, Greenhalgh MD, Stead D, Ng S, Slawin AMZ, Smith AD. Isothiourea-Catalyzed Enantioselective α-Alkylation of Esters via 1,6-Conjugate Addition to para-Quinone Methides. Molecules. 2021; 26(21):6333. https://doi.org/10.3390/molecules26216333
Chicago/Turabian StyleArokianathar, Jude N., Will C. Hartley, Calum McLaughlin, Mark D. Greenhalgh, Darren Stead, Sean Ng, Alexandra M. Z. Slawin, and Andrew D. Smith. 2021. "Isothiourea-Catalyzed Enantioselective α-Alkylation of Esters via 1,6-Conjugate Addition to para-Quinone Methides" Molecules 26, no. 21: 6333. https://doi.org/10.3390/molecules26216333
APA StyleArokianathar, J. N., Hartley, W. C., McLaughlin, C., Greenhalgh, M. D., Stead, D., Ng, S., Slawin, A. M. Z., & Smith, A. D. (2021). Isothiourea-Catalyzed Enantioselective α-Alkylation of Esters via 1,6-Conjugate Addition to para-Quinone Methides. Molecules, 26(21), 6333. https://doi.org/10.3390/molecules26216333