
Discovery of New Ginsenol-Like Compounds with High Antiviral Activity


 ,
,  , , , ,
, , , ,  ,
,  and
and
Abstract
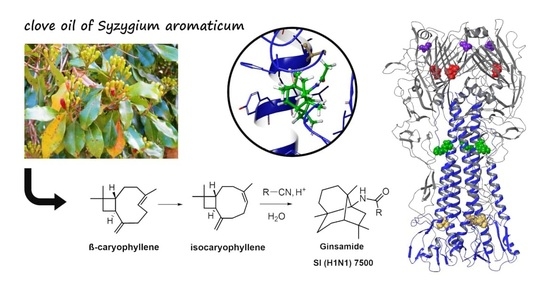
:
1. Introduction
2. Results and Discussion
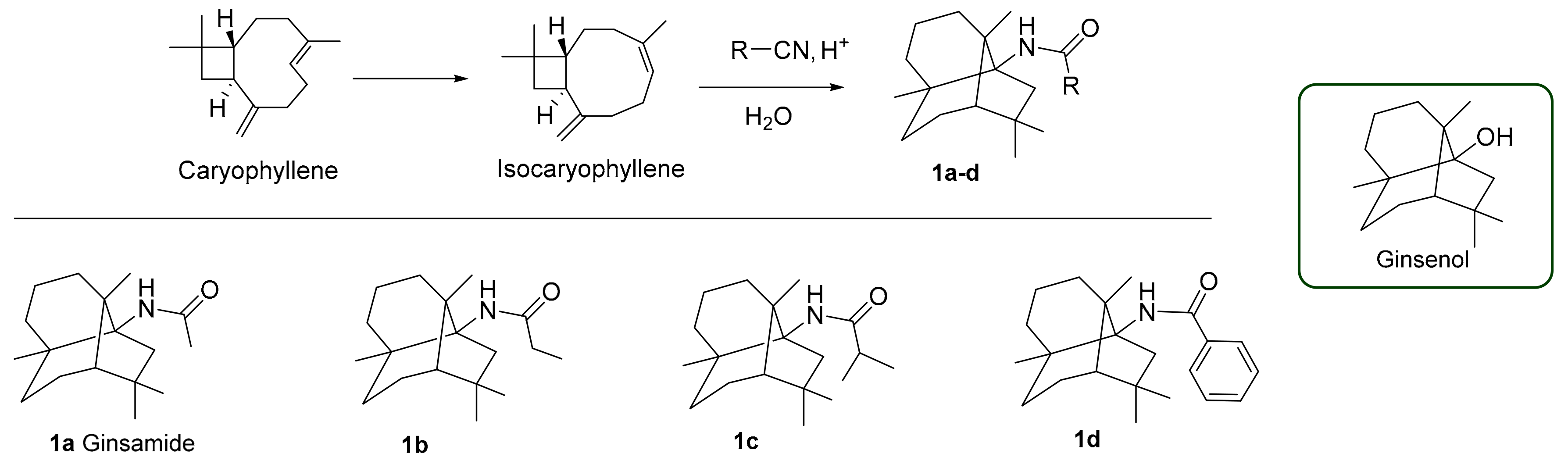
2.1. Chemistry
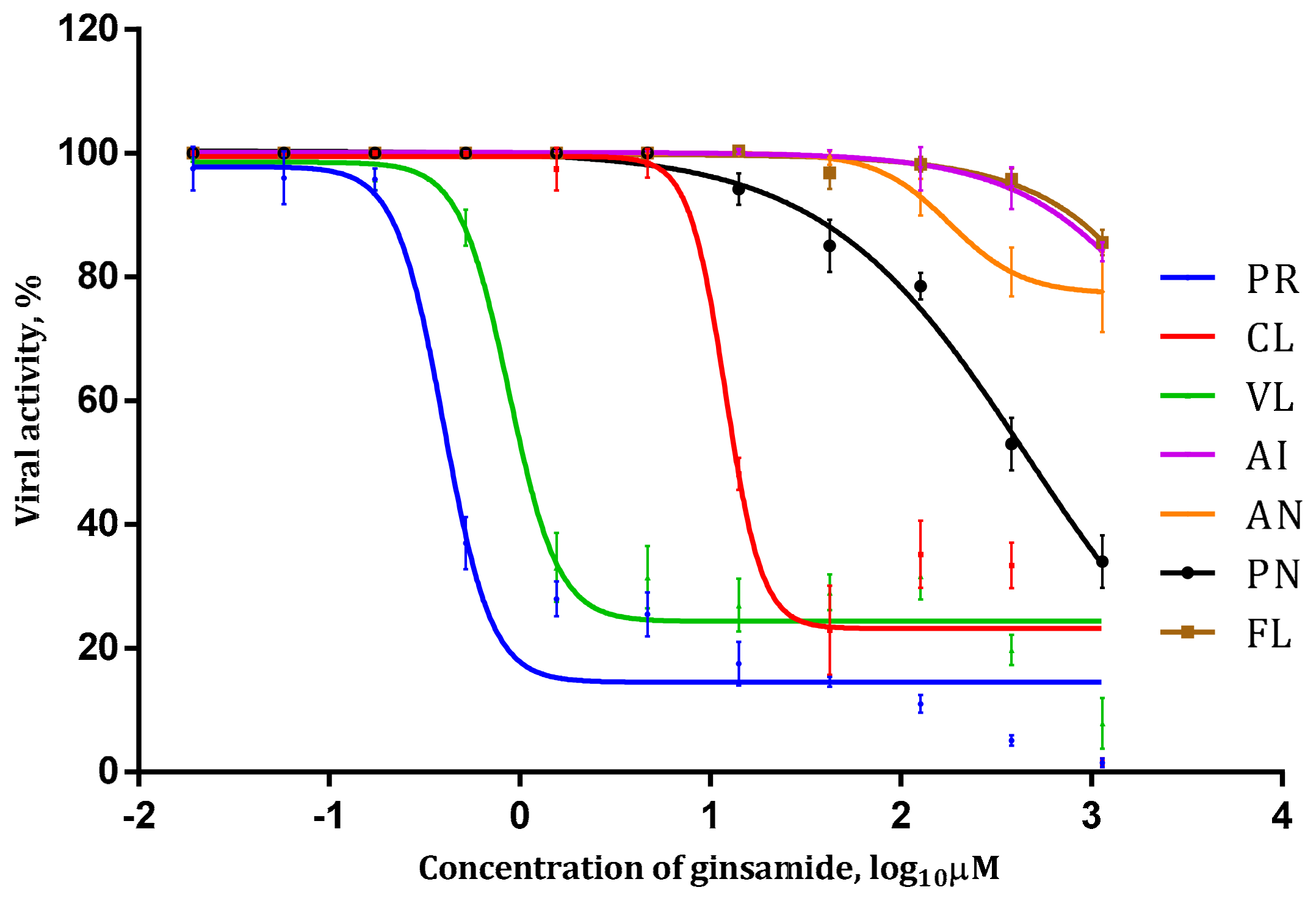
2.2. Antiviral Activity Study
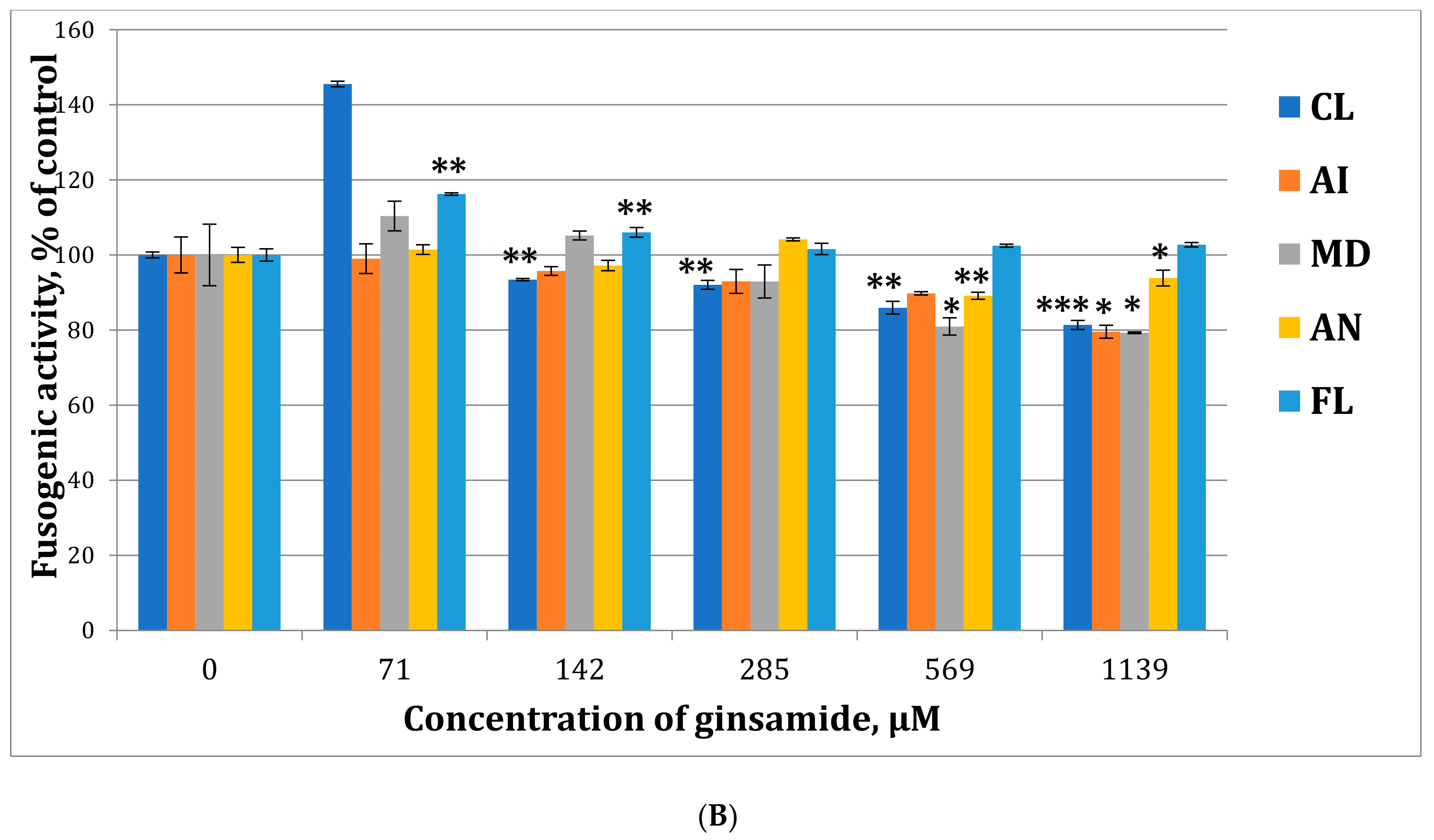
2.3. A Study of the Mechanism of Antiviral Activity
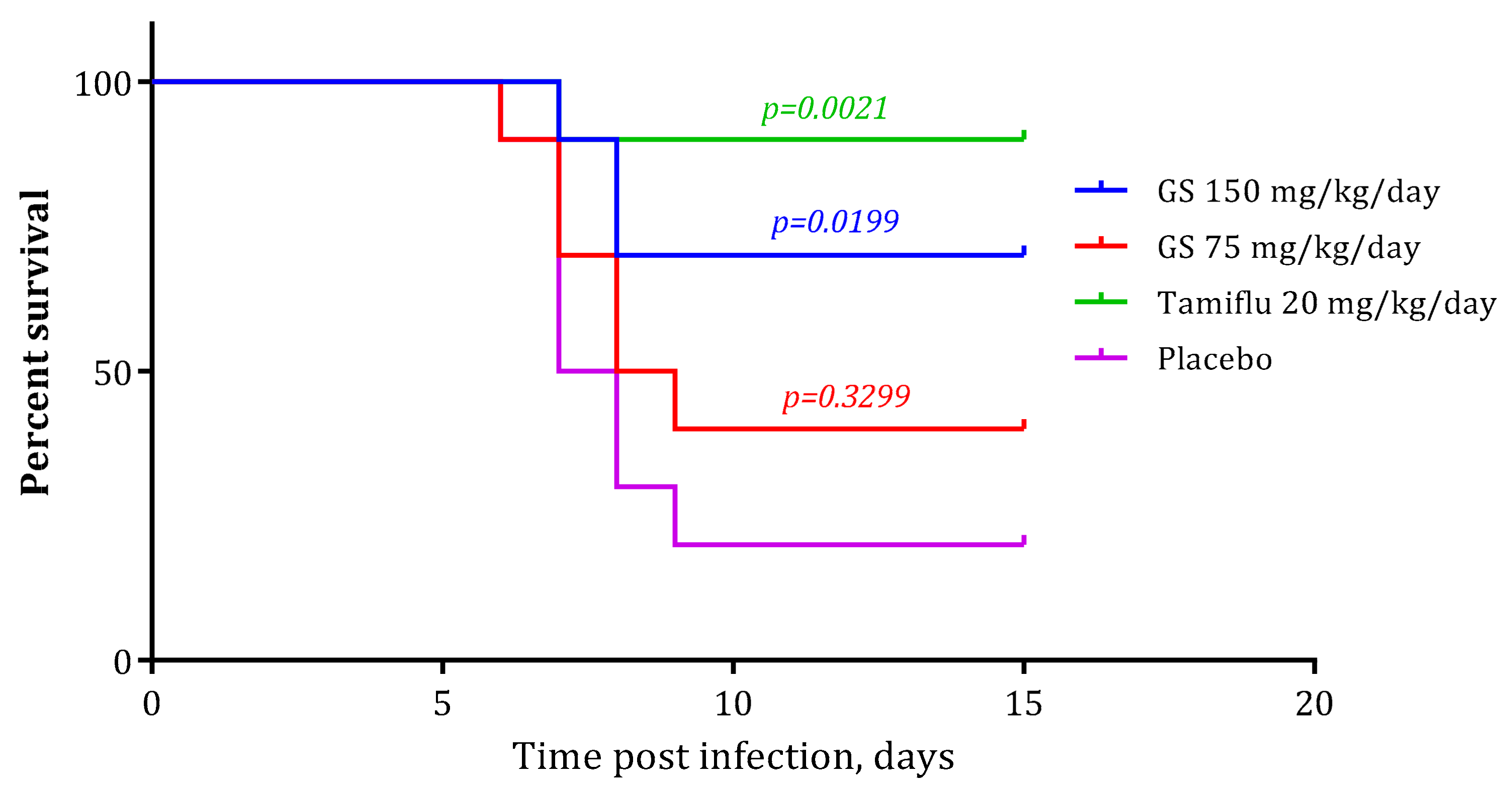
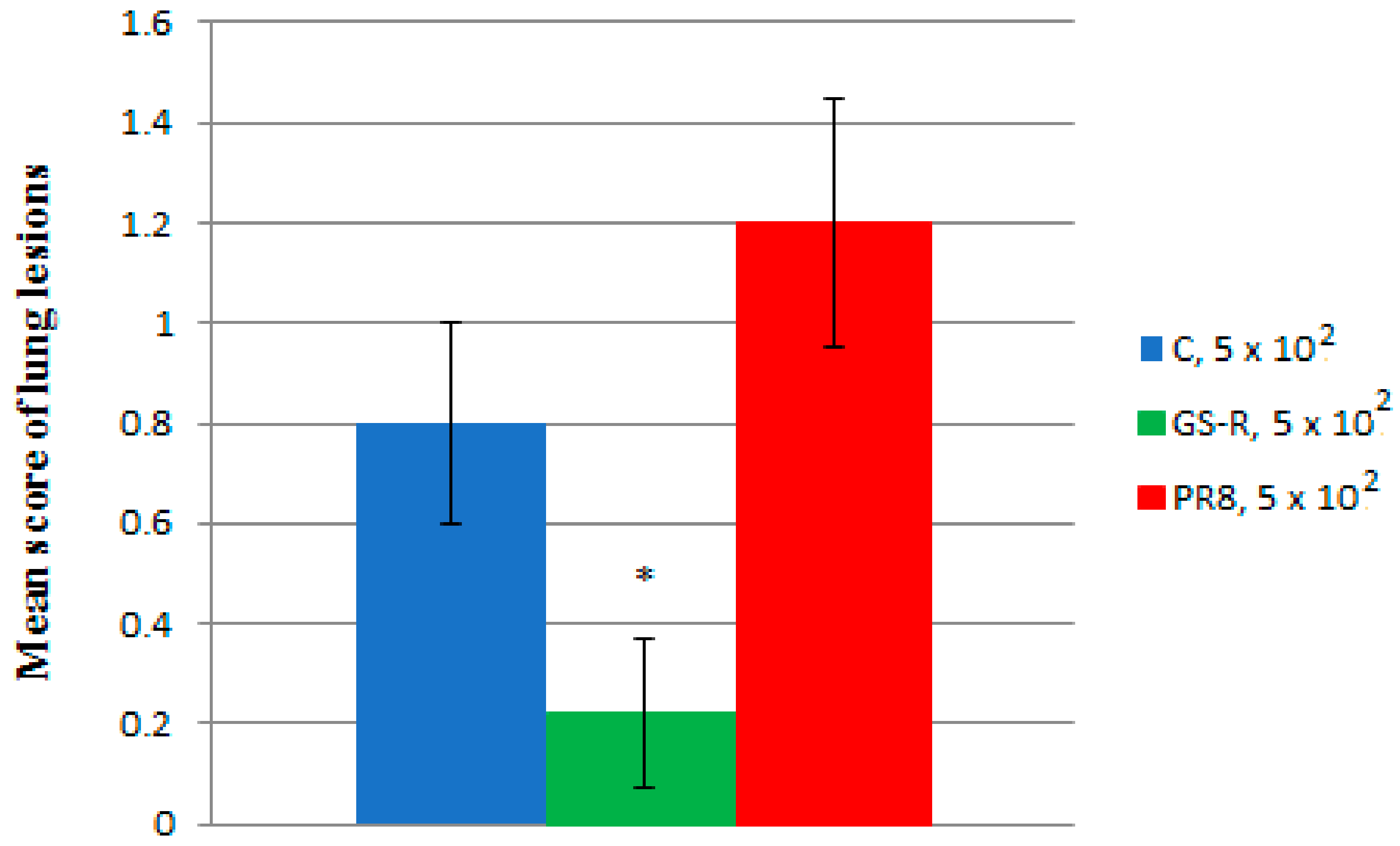
2.4. In Vivo Experiments
2.5. Propagation with Ginsamide in Cell Culture Results in Selection of GS-Resistant Strains of Influenza A Virus
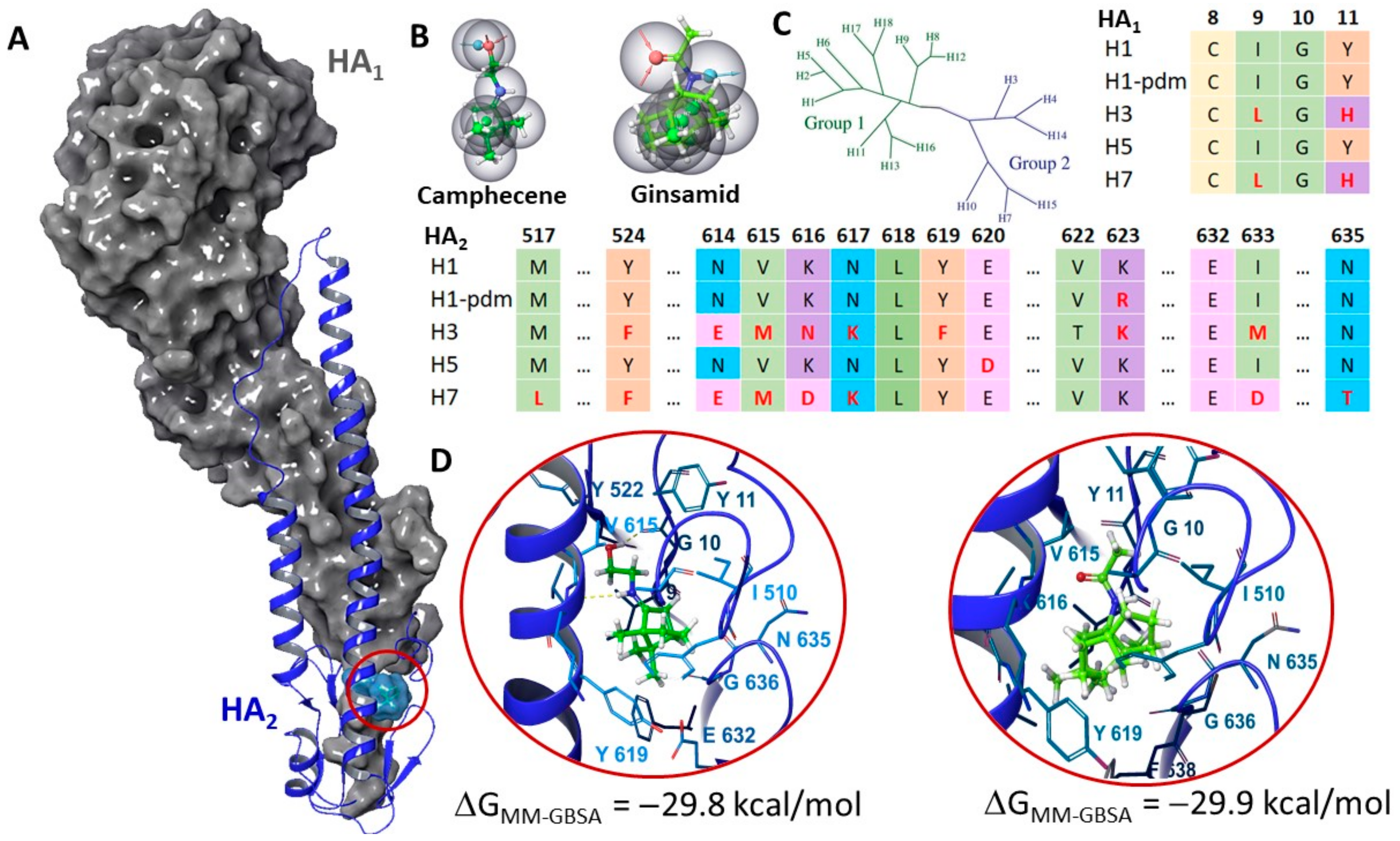
2.6. Molecular Modeling Study
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Procedure for Synthesis of Derivatives 1b–d
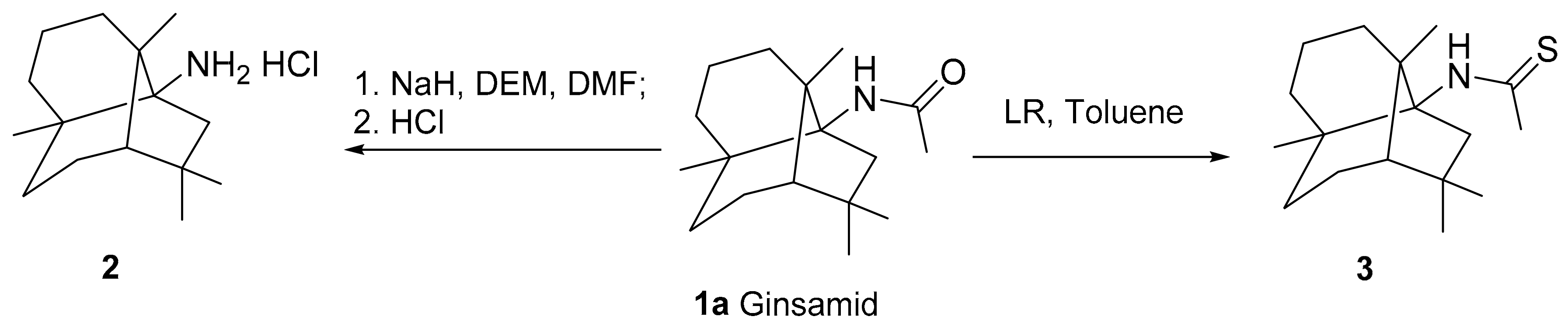
3.1.3. Synthesis of an Amine 2 from Ginsamide
3.1.4. Synthesis of a Thioamide 3a
3.2. Biological Studies
3.2.1. Viruses and Cells
3.2.2. Animals
3.2.3. Cytotoxicity Assay
3.2.4. CPE Reduction Assay
3.2.5. Virus Titration and Virus Yield Reduction Assay
3.2.6. Time-of-Addition Experiments
3.2.7. Haemolysis Assay
3.2.8. Hemagglutination Inhibition Assay
3.2.9. In Vitro Selection and Analysis of Resistant Mutants
3.2.10. In Vivo Experiments
3.2.11. Statistical Analysis
3.3. Computation Details
3.3.1. Protein and Ligand Preparation
3.3.2. Bindin Site Analysis
3.3.3. Molecular Docking Procedure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Viasus, D.; Revuelta, J.A.O.; Martínez-Montauti, J.; Carratalà, J. Influenza A(H1N1)pdm09-related pneumonia and other complications. Enferm. Infecc. Microbiol. Clin. 2012, 30, 43–48. [Google Scholar] [CrossRef]
- Vousden, N.; Knight, M. Lessons learned from the A (H1N1) influenza pandemic. Best Pract. Res. Clin. Obstet. Gynaecol. 2020, 76, 41–52. [Google Scholar] [CrossRef]
- Karlsson, E.A.; Marcelin, G.; Webby, R.J.; Schultz-Cherry, S. Review on the impact of pregnancy and obesity on influenza virus infection. Influenza Other Respir. Viruses 2012, 6, 449–460. [Google Scholar] [CrossRef]
- Philippon, D.A.M.; Wu, P.; Cowling, B.J.; Lau, E.H.Y. Avian Influenza Human Infections at the Human-Animal Interface. J. Infect. Dis. 2020, 222, 528–537. [Google Scholar] [CrossRef]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane Resistance Among Influenza A Viruses Isolated Early during the 2005–2006 Influenza Season in the United States. JAMA 2006, 295, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, E.; Ye, D.; Li, J.; Zhang, D.; Wang, J.; Zhao, F.; Hilgenfeld, R.; Zheng, M.; Jiang, H.; Liu, H. Recent Advances in Neuraminidase Inhibitor Development as Anti-influenza Drugs. ChemMedChem 2012, 7, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Peramivir: A Review in Uncomplicated Influenza. Drugs 2018, 78, 1363–1370. [Google Scholar] [CrossRef]
- Kashiwagi, S.; Yoshida, S.; Yamaguchi, H.; Niwa, S.; Mitsui, N.; Tanigawa, M.; Shiosakai, K.; Yamanouchi, N.; Shiozawa, T.; Yamaguchi, F. Safety of the long-acting neuraminidase inhibitor laninamivir octanoate hydrate in post-marketing surveillance. Int. J. Antimicrob. Agents 2012, 40, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Sugaya, N.; Hirotsu, N.; Lee, N.; de Jong, M.D.; Hurt, A.C.; Ishida, T.; Sekino, H.; Yamada, K.; Portsmouth, S.; et al. Baloxavir Marboxil for Uncomplicated Influenza in Adults and Adolescents. N. Engl. J. Med. 2018, 379, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, S.; Tiezzi, A.; Masci, V.L.; Ovidi, E. Natural products for human health: An historical overview of the drug discovery approaches. Nat. Prod. Res. 2018, 32, 1926–1950. [Google Scholar] [CrossRef]
- Salakhutdinov, N.F.; Volcho, K.P.; Yarovaya, O.I. Monoterpenes as a renewable source of biologically active compounds. Pure Appl. Chem. 2017, 89, 1105–1117. [Google Scholar] [CrossRef]
- Balunas, M.J.; Kinghorn, A.D. Drug discovery from medicinal plants. Life Sci. 2005, 78, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Rishton, G.M. Natural Products as a Robust Source of New Drugs and Drug Leads: Past Successes and Present Day Issues. Am. J. Cardiol. 2008, 101, S43–S49. [Google Scholar] [CrossRef] [PubMed]
- Yarovaya, O.I.; Salakhutdinov, N.F. Mono- and sesquiterpenes as a starting platform for the development of antiviral drugs. Russ. Chem. Rev. 2021, 90, 488–510. [Google Scholar] [CrossRef]
- Zielińska-Błajet, M.; Feder-Kubis, J. Monoterpenes and their derivatives—Recent development in biological and medical applications. Int. J. Mol. Sci. 2020, 21, 7078. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Cui, Q.; Caffrey, M.; Rong, L.; Du, R. Small molecule inhibitors of influenza virus entry. Pharmaceuticals 2021, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Yarovaya, O.I.; Baranova, D.V.; Galochkina, A.V.; Shtro, A.A.; Kireeva, M.V.; Borisevich, S.S.; Gatilov, Y.V.; Zarubaev, V.V.; Salakhutdinov, N.F. Quaternary ammonium salts based on (−)-borneol as effective inhibitors of influenza virus. Arch. Virol. 2021, 166, 1965–1976. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Baev, D.S.; Shernyukov, A.V.; Shtro, A.A.; Zarubaev, V.V.; Salakhutdinov, N.F. Aliphatic and alicyclic camphor imines as effective inhibitors of influenza virus H1N1. Eur. J. Med. Chem. 2017, 127, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Zarubaev, V.V.; Garshinina, A.V.; Tretiak, T.S.; Fedorova, V.A.; Shtro, A.A.; Sokolova, A.S.; Yarovaya, O.I.; Salakhutdinov, N.F. Broad range of inhibiting action of novel camphor-based compound with anti-hemagglutinin activity against influenza viruses in vitro and in vivo. Antivir. Res. 2015, 120, 126–133. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakova, N.S.; Zaykovskaya, A.V.; Baev, D.S.; Tolstikova, T.G.; Shcherbakov, D.N.; Pyankov, O.V.; et al. Monoterpenoid-based inhibitors of filoviruses targeting the glycoprotein-mediated entry process. Eur. J. Med. Chem. 2020, 207, 112726. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.S.; Putilova, V.P.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; Shcherbakov, D.N.; Orshanskaya, I.R.; Sinegubova, E.O.; Esaulkova, I.L.; et al. Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses. Molecules 2021, 26, 2235. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Kovaleva, K.S.; Yarovaya, O.I.; Bormotov, N.I.; Shishkina, L.N.; Serova, O.A.; Sergeev, A.A.; Agafonov, A.P.; Maksuytov, R.A.; Salakhutdinov, N.F. (+)-Camphor and (−)-borneol derivatives as potential anti-orthopoxvirus agents. Arch. Pharm. 2021, 354, e2100038. [Google Scholar] [CrossRef] [PubMed]
- Yarovaya, O.I.; Kovaleva, K.S.; Zaykovskaya, A.A.; Yashina, L.N.; Scherbakova, N.S.; Scherbakov, D.N.; Borisevich, S.S.; Zubkov, F.I.; Antonova, A.S.; Peshkov, R.Y.; et al. New class of hantaan virus inhibitors based on conjugation of the isoindole fragment to (+)-camphor or (−)-fenchone hydrazonesv. Bioorg. Med. Chem. Lett. 2021, 40, 127926. [Google Scholar] [CrossRef] [PubMed]
- Astani, A.; Reichling, J.; Schnitzler, P. Screening for Antiviral Activities of Isolated Compounds from Essential Oils. Evid. Based Complement. Altern. Med. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Peng, C.; Xie, X.; Zhang, S.; Cao, X. Availability, Pharmaceutics, Security, Pharmacokinetics, and Pharmacological Activities of Patchouli Alcohol. Evid. Based Complement. Altern. Med. 2017, 2017, 1–9. [Google Scholar] [CrossRef]
- Wu, H.; Li, B.; Wang, X.; Jin, M.; Wang, G. Inhibitory Effect and Possible Mechanism of Action of Patchouli Alcohol against Influenza A (H2N2) Virus. Molecules 2011, 16, 6489–6501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, G.-Q.; Kenney, P.M.; Lam, L.K.T. Sesquiterpenes from Clove (Eugenia caryophyllata) as Potential Anticarcinogenic Agents. J. Nat. Prod. 1992, 55, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Sylvestre, M.; Legault, J.; Dufour, D.; Pichette, A. Chemical composition and anticancer activity of leaf essential oil of Myrica gale L. Phytomedicine 2005, 12, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Sylvestre, M.; Pichette, A.; Lavoie, S.; Longtin, A.; Legault, J. Composition and cytotoxic activity of the leaf essential oil of Comptonia peregrina (L.) Coulter. Phytother. Res. 2007, 21, 536–540. [Google Scholar] [CrossRef]
- Goren, A.C.; Piozzi, F.; Akcicek, E.; Kılıç, T.; Çarıkçı, S.; Mozioğlu, E.; Setzer, W.N. Essential oil composition of twenty-two Stachys species (mountain tea) and their biological activities. Phytochem. Lett. 2011, 4, 448–453. [Google Scholar] [CrossRef]
- Sharma, C.; Sadek, B.; Goyal, S.N.; Sinha, S.; Kamal, M.A.; Ojha, S. Small Molecules from Nature Targeting G-Protein Coupled Cannabinoid Receptors: Potential Leads for Drug Discovery and Development. Evid. Based Complement. Altern. Med. 2015, 2015, 1–26. [Google Scholar] [CrossRef]
- Ghelardini, C.; Galeotti, N.; Mannelli, L.D.C.; Mazzanti, G.; Bartolini, A. Local anaesthetic activity of β-caryophyllene. Il Farm. 2001, 56, 387–389. [Google Scholar] [CrossRef]
- Fidyt, K.; Fiedorowicz, A.; Strządała, L.; Szumny, A. β-caryophyllene and β-caryophyllene oxide-natural compounds of anticancer and analgesic properties. Cancer Med. 2016, 5, 3007–3017. [Google Scholar] [CrossRef]
- Legault, J.; Pichette, A. Potentiating effect of β-caryophyllene on anticancer activity of α-humulene, isocaryophyllene and paclitaxel. J. Pharm. Pharmacol. 2010, 59, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Neta, M.C.S.; Vittorazzi, C.; Guimarães, A.C.; Martins, J.D.L.; Fronza, M.; Endringer, D.C.; Scherer, R. Effects of β-caryophyllene and Murraya paniculata essential oil in the murine hepatoma cells and in the bacteria and fungi 24-h time–kill curve studies. Pharm. Biol. 2017, 55, 190–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitjer, L.; Malich, A.; Paschke, C.; Kluge, S.; Gerke, R.; Rissom, B.; Weiser, J.; Noltemeyer, M. Rearrangement of (-)-β-Caryophyllene. A Product Analysis and Force Field Study. J. Am. Chem. Soc. 1995, 117, 9180–9189. [Google Scholar] [CrossRef]
- Yarovaya, O.I.; Korchagina, D.V.; Rybalova, T.V.; Gatilov, Y.V.; Polovinka, M.P.; Barkhash, V.A. Reactions of caryophyllene, isocaryophyllene, and their epoxy derivatives with acetonitrile under Ritter reaction conditions. Russ. J. Org. Chem. 2004, 40, 1593–1598. [Google Scholar] [CrossRef]
- Chen, M.-E.; Chen, X.-W.; Hu, Y.-H.; Ye, R.; Lv, J.-W.; Li, B.; Zhang, F.-M. Recent advances of Ritter reaction and its synthetic applications. Org. Chem. Front. 2021, 8, 4623–4664. [Google Scholar] [CrossRef]
- Ziarani, G.M.; Hasankiadeh, F.S.; Mohajer, F. Recent Applications of Ritter Reactions in Organic Syntheses. ChemistrySelect 2020, 5, 14349–14379. [Google Scholar] [CrossRef]
- Bolsakova, J.; Jirgensons, A. The Ritter reaction for the synthesis of heterocycles. Chem. Heterocycl. Compd. 2017, 53, 1167–1177. [Google Scholar] [CrossRef]
- Cho, I.H. Volatile compounds of ginseng (Panax sp.): A review. J. Korean Soc. Appl. Biol. Chem. 2015, 58, 67–75. [Google Scholar] [CrossRef]
- Salomatina, O.V.; Yarovaya, O.I.; Korchagina, D.V.; Gatilov, Y.V.; Polovinka, M.P.; Barkhash, V.A. Transformations of isocaryophyllene diepoxide under conditions of homogeneous and heterogeneous acid catalysis. Russ. J. Org. Chem. 2005, 41, 1280–1285. [Google Scholar] [CrossRef]
- Zarubaev, V.V.; Pushkina, E.A.; Borisevich, S.S.; Galochkina, A.V.; Garshinina, A.V.; Shtro, A.A.; Egorova, A.A.; Sokolova, A.S.; Khursan, S.L.; Yarovaya, O.I.; et al. Selection of influenza virus resistant to the novel camphor-based antiviral camphecene results in loss of pathogenicity. Virology 2018, 524, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Scholtissek, C.; Quack, G.; Klenk, H.; Webster, R. How to overcome resistance of influenza A viruses against adamantane derivatives. Antivir. Res. 1998, 37, 83–95. [Google Scholar] [CrossRef]
- Hoopes, J.D.; Driebe, E.M.; Kelley, E.; Engelthaler, D.M.; Keim, P.S.; Perelson, A.S.; Rong, L.; Went, G.T.; Nguyen, J.T. Triple Combination Antiviral Drug (TCAD) Composed of Amantadine, Oseltamivir, and Ribavirin Impedes the Selection of Drug-Resistant Influenza A Virus. PLoS ONE 2011, 6, e29778. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.; Kadowaki, S.; Watanabe, I.; Kadowaki, Y.; Takei, M.; Fukuda, H. PA subunit of RNA polymerase as a promising target for anti-influenza virus agents. Antivir. Res. 2008, 78, 194–201. [Google Scholar] [CrossRef]
- Russell, R.J.; Kerry, P.S.; Stevens, D.J.; Steinhauer, D.A.; Martin, S.R.; Gamblin, S.J.; Skehel, J.J. Structure of influenza hemagglutinin in complex with an inhibitor of membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 17736–17741. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, Y.; Igarashi, M.; Okamatsu, M.; Noshi, T.; Sakoda, Y.; Yamamoto, N.; Ito, K.; Yoshida, R.; Kida, H. Antiviral activity of stachyflin on influenza A viruses of different hemagglutinin subtypes. Virol. J. 2013, 10, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagita, H.; Yamamoto, N.; Fuji, H.; Liu, X.; Ogata, M.; Yokota, M.; Takaku, H.; Hasegawa, H.; Odagiri, T.; Tashiro, M.; et al. Mechanism of drug resistance of hemagglutinin of influenza virus and potent scaffolds inhibiting its function. ACS Chem. Biol. 2012, 7, 552–562. [Google Scholar] [CrossRef]
- Tang, G.; Lin, X.; Qiu, Z.; Li, W.; Zhu, L.; Wang, L.; Li, S.; Li, H.; Lin, W.; Yang, M.; et al. Design and synthesis of benzenesulfonamide derivatives as potent anti-influenza hemagglutinin inhibitors. ACS Med. Chem. Lett. 2011, 2, 603–607. [Google Scholar] [CrossRef]
- Combrink, K.D.; Gulgeze, H.B.; Yu, K.L.; Pearce, B.C.; Trehan, A.K.; Wei, J.; Deshpande, M.; Krystal, M.; Torri, A.; Luo, G.; et al. Salicylamide inhibitors of influenza virus fusion. Bioorg. Med. Chem. Lett. 2000, 10, 1649–1652. [Google Scholar] [CrossRef]
- Plotch, S.J.; O’Hara, B.; Morin, J.; Palant, O.; LaRocque, J.; Bloom, J.D.; Lang, S.A.; DiGrandi, M.J.; Bradley, M.; Nilakantan, R.; et al. Inhibition of Influenza A Virus Replication by Compounds Interfering with the Fusogenic Function of the Viral Hemagglutinin. J. Virol. 1999, 73, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, B.; Chang, G.; Hu, Y.; Zhan, D.; Xia, Y.; Li, Y.; Yang, Y.; Zhu, Q. Virulence of H5N1 virus in mice attenuates after in vitro serial passages. Virol. J. 2011, 8, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisevich, S.S.; Gureev, M.A.; Yarovaya, O.I.; Zarubaev, V.V.; Kostin, G.A.; Porozov, Y.B.; Salakhutdinov, N.F. Can molecular dynamics explain decreased pathogenicity in mutant camphecene-resistant influenza virus? J. Biomol. Struct. Dyn. 2021, 1–12. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The structure and receptor binding properties of the 1918 influenza hemagglutinin. Science 2004, 303, 1838–1842. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolova, A.S.; Yarovaya, O.I.; Shernyukov, A.V.; Gatilov, Y.V.; Razumova, Y.V.; Zarubaev, V.V.; Tretiak, T.S.; Pokrovsky, A.G.; Kiselev, O.I.; Salakhutdinov, N.F. Discovery of a new class of antiviral compounds: Camphor imine derivatives. Eur. J. Med. Chem. 2015, 105, 263–273. [Google Scholar] [CrossRef]
- Xu, R.; Ekiert, D.C.; Krause, J.C.; Hai, R.; Crowe, J.E.; Wilson, I.A. Structural Basis of Preexisting Immunity to the 2009 H1N1 Pandemic Influenza Virus. Science 2010, 328, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Kadam, R.U.; Wilson, I.A. A small-molecule fragment that emulates binding of receptor and broadly neutralizing antibodies to influenza A hemagglutinin. Proc. Natl. Acad. Sci. USA 2018, 115, 4240–4245. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Peng, R.; Zhang, W.; Qi, J.; Song, H.; Liu, S.; Wang, H.; Wang, M.; Xiao, H.; Fu, L.; et al. Avian-to-Human Receptor-Binding Adaptation of Avian H7N9 Influenza Virus Hemagglutinin. Cell Rep. 2019, 29, 2217–2228.e5. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Maeda, T.; Ohnishi, S. Activation of influenza virus by acidic media causes hemolysis and fusion of erythrocytes. FEBS Lett. 1980, 122, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.A.; Hu, Y.; Yamamoto, Y.; Hoe, N.B.; Wei, T.S.; Mu, D.; Sun, Y.; Joo, L.S.; Dagher, R.; Zielonka, E.M.; et al. Distal Airway Stem Cells Yield Alveoli In Vitro and during Lung Regeneration following H1N1 Influenza Infection. Cell 2011, 147, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]















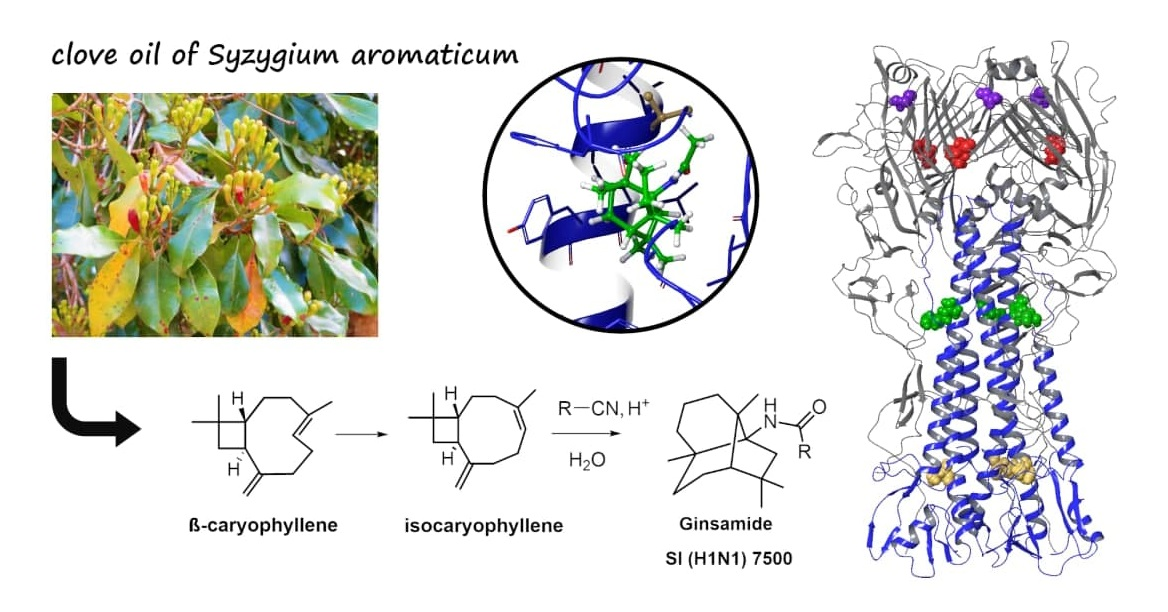{kind=link}
{kind=link}
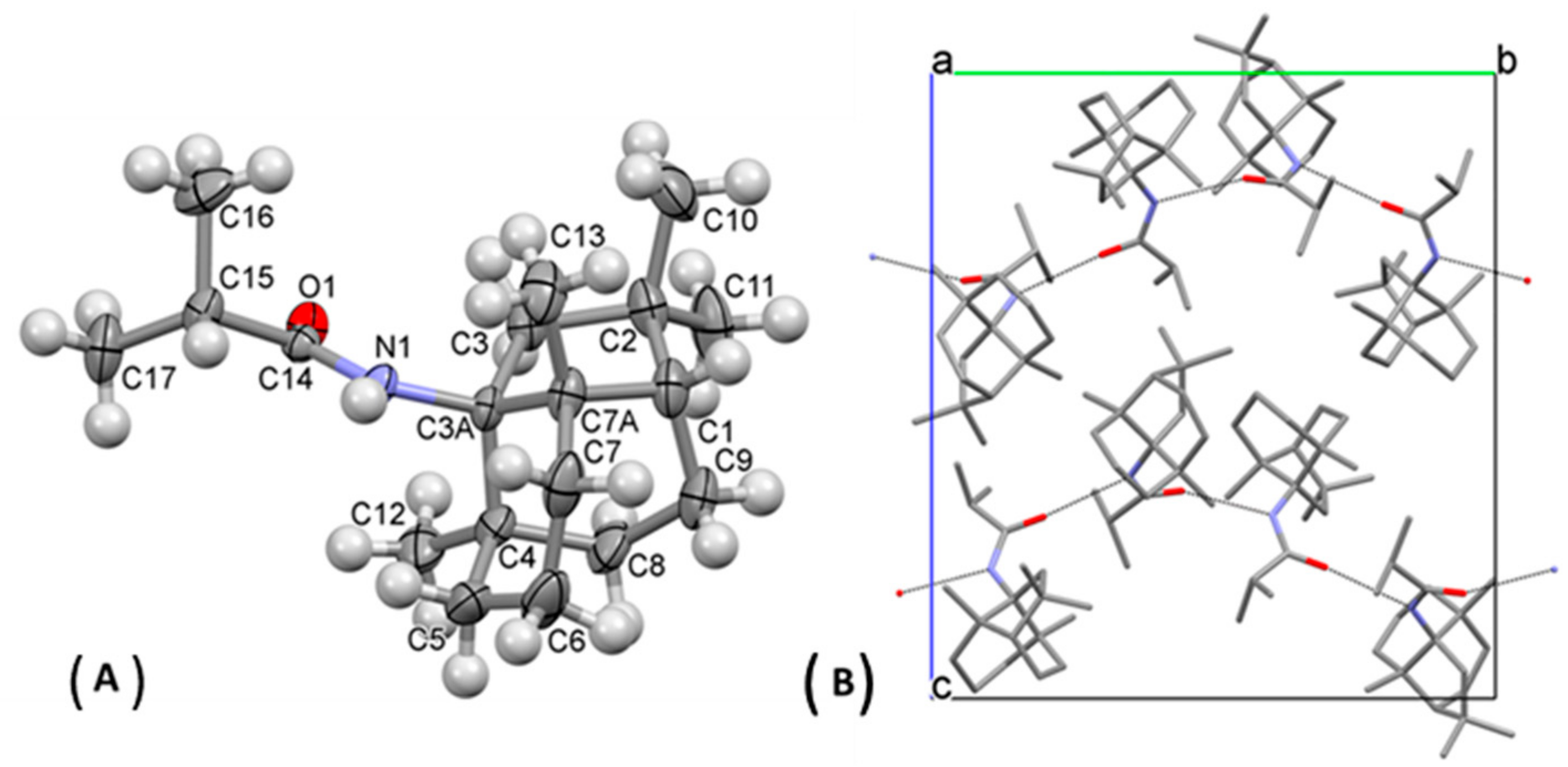{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CC50 (µM) a | IC50 H1N1 (µM) b | SI c |
|---|---|---|---|
| 1a | >1140 | 0.152 ± 0.03 | 7500 |
| 1b | 128.5 ± 9.4 | 0.144 ± 0.019 | 890 |
| 1c | >1031 | 0.21 ± 0.029 | 5000 |
| 1d | 141.3 ± 11.5 | 9.2 ± 1.1 | 15 |
| 2 | 15.4 ± 1.2 | 0.45 ± 0.06 | 34 |
| 3 | >1126 | 1.13 ± 0.19 | 1000 |
| Oseltamivir | >200 | 0.31 ± 0.04 | 645 |
| Ribavirin | >2000 | 24.6 ± 3.7 | >81 |
| Rimantadine | 335 ± 27 | 67.0 ± 4.9 | 5 |
| Virus | CC50 (µM) a | IC50 H1N1 (µM) b | SI c |
|---|---|---|---|
| A/Puerto Rico/8/1934 (H1N1) | >1140 | 0.152 ± 0.03 | 7500 |
| A/California/7/2009 (H1N1) pdm09 | >1140 | 10.65 ± 1.3 | 107 |
| A/Vladivostok/2/2009 (H1N1) | >1140 | 0.38 ± 0.051 | 3000 |
| A/Aichi/2/1968 (H3N2) | >1140 | 789 ± 82 | 15 |
| A/Anhui/1/2013 (H7N9) | >1140 | 125.5 ± 11.8 | 9 |
| A/Mallard/Pennsylvania/1984 (H5N2) | >1140 | 133.1 ± 15.6 | 9 |
| B/Florida/4/2006 (Yamagata-like) | >1140 | 1125.5 ± 141.4 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volobueva, A.S.; Yarovaya, O.I.; Kireeva, M.V.; Borisevich, S.S.; Kovaleva, K.S.; Mainagashev, I.Y.; Gatilov, Y.V.; Ilyina, M.G.; Zarubaev, V.V.; Salakhutdinov, N.F. Discovery of New Ginsenol-Like Compounds with High Antiviral Activity. Molecules 2021, 26, 6794. https://doi.org/10.3390/molecules26226794
Volobueva AS, Yarovaya OI, Kireeva MV, Borisevich SS, Kovaleva KS, Mainagashev IY, Gatilov YV, Ilyina MG, Zarubaev VV, Salakhutdinov NF. Discovery of New Ginsenol-Like Compounds with High Antiviral Activity. Molecules. 2021; 26(22):6794. https://doi.org/10.3390/molecules26226794
Chicago/Turabian StyleVolobueva, Aleksandrina S., Olga I. Yarovaya, Marina V. Kireeva, Sophia S. Borisevich, Kseniya S. Kovaleva, Iliya Ya. Mainagashev, Yuri V. Gatilov, Margarita G. Ilyina, Vladimir V. Zarubaev, and Nariman F. Salakhutdinov. 2021. "Discovery of New Ginsenol-Like Compounds with High Antiviral Activity" Molecules 26, no. 22: 6794. https://doi.org/10.3390/molecules26226794
APA StyleVolobueva, A. S., Yarovaya, O. I., Kireeva, M. V., Borisevich, S. S., Kovaleva, K. S., Mainagashev, I. Y., Gatilov, Y. V., Ilyina, M. G., Zarubaev, V. V., & Salakhutdinov, N. F. (2021). Discovery of New Ginsenol-Like Compounds with High Antiviral Activity. Molecules, 26(22), 6794. https://doi.org/10.3390/molecules26226794