
Toward a Disease-Modifying Therapy of Alpha-Synucleinopathies: New Molecules and New Approaches Came into the Limelight


Abstract
:1. Introduction
1.1. Pathological Aggregation of Alpha-Synuclein
1.2. The Neuroanatomical and Cell-to-Cell Propagation of Alpha-Synuclein Aggregation Pathology
1.3. Pathological Consequences of the Accumulation of Products of Alpha-Synuclein Aggregation
2. Post-Transcriptional Targeting of Alpha-Synuclein Production
2.1. Strategies to Achieve Post-Transcriptional Alpha-Synuclein Knockdown
2.2. Methods of Drug Delivery
3. Immunotherapeutic Approaches for Alpha-Synuclein Degradation
3.1. Active Immunization
3.2. Passive Immunization
4. Current Limitations, Challenges and Future Directions
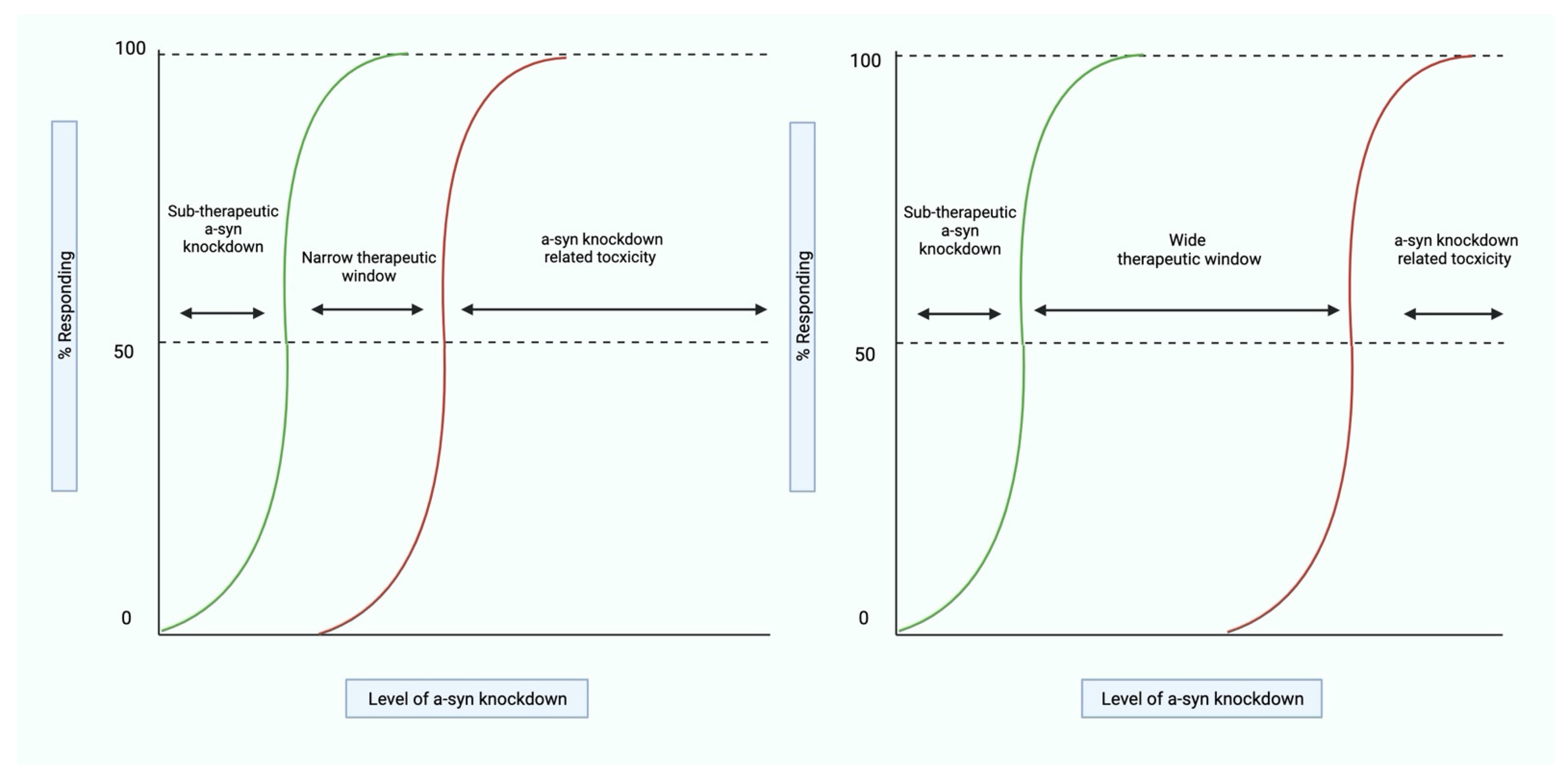
4.1. Establishing the Therapeutic Window for Alpha-Synuclein Knockdown
4.2. Limitations Specific to Immunotherapies
4.3. Optimizing Preclinical Data
4.4. Enhancing Clinical Trial Design
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goedert, M.; Jakes, R.; Spillantini, M.G. The synucleinopathies: Twenty years on. J. Parkinson’s Dis. 2017, 7, S53–S71. [Google Scholar] [CrossRef] [Green Version]
- Surguchov, A. Intracellular dynamics of synucleins: “Here, there and everywhere”. Int. Rev. Cell Mol. Biol 2015, 320, 103–169. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar] [CrossRef]
- Uversky, V.N. Looking at the recent advances in understanding alpha-synuclein and its aggregation through the proteoform prism. F1000Res 2017, 6, 525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burre, J.; Sharma, M.; Sudhof, T.C. Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A. Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity? Curr. Opin. Chem. Biol. 2021, 64, 67–75. [Google Scholar] [CrossRef]
- Fares, M.B.; Jagannath, S.; Lashuel, H.A. Reverse engineering Lewy bodies: How far have we come and how far can we go? Nat. Rev. Neurosci. 2021, 22, 111–131. [Google Scholar] [CrossRef]
- Braak, H.; del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horvath-Puho, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sorensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Parkkinen, L.; Kauppinen, T.; Pirttila, T.; Autere, J.M.; Alafuzoff, I. Alpha-synuclein pathology does not predict extrapyramidal symptoms or dementia. Ann. Neurol. 2005, 57, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R. Prying into the Prion hypothesis for Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 9808–9818. [Google Scholar] [CrossRef]
- Watts, J.C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D.T.; Gentleman, S.M.; DeArmond, S.J.; Prusiner, S.B. Transmission of multiple system atrophy prions to transgenic mice. Proc. Natl. Acad. Sci. USA 2013, 110, 19555–19560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacino, A.N.; Brooks, M.; McKinney, A.B.; Thomas, M.A.; Shaw, G.; Golde, T.E.; Giasson, B.I. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 12368–12378. [Google Scholar] [CrossRef] [Green Version]
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Mougenot, A.L.; Nicot, S.; Bencsik, A.; Morignat, E.; Verchere, J.; Lakhdar, L.; Legastelois, S.; Baron, T. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging 2012, 33, 2225–2228. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain J. Neurol. 2013, 136, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, A.R.; Ninkina, N.N.; Hanger, D.P.; Anderton, B.H.; Davies, A.M.; Buchman, V.L. Induction of neuronal death by alpha-synuclein. Eur. J. Neurosci. 2000, 12, 3073–3077. [Google Scholar] [CrossRef]
- Buchman, V.L.; Ninkina, N. Modulation of alpha-synuclein expression in transgenic animals for modelling synucleinopathies—Is the juice worth the squeeze? Neurotox. Res. 2008, 14, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Al-Wandi, A.; Ninkina, N.; Millership, S.; Williamson, S.J.; Jones, P.A.; Buchman, V.L. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 2010, 31, 796–804. [Google Scholar] [CrossRef] [Green Version]
- Senior, S.L.; Ninkina, N.; Deacon, R.; Bannerman, D.; Buchman, V.L.; Cragg, S.J.; Wade-Martins, R. Increased striatal dopamine release and hyperdopaminergic-like behaviour in mice lacking both alpha-synuclein and gamma-synuclein. Eur. J. Neurosci. 2008, 27, 947–957. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, K.; Sinha, M.; Pham Cle, L.; Jana, S.; Chanda, D.; Cappai, R.; Chakrabarti, S. Alpha-synuclein induced membrane depolarization and loss of phosphorylation capacity of isolated rat brain mitochondria: Implications in Parkinson’s disease. FEBS Lett. 2010, 584, 1571–1576. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.H.; Cristovao, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.S. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricke, K.M.; Pass, T.; Kimoloi, S.; Fahrmann, K.; Jungst, C.; Schauss, A.; Baris, O.R.; Aradjanski, M.; Trifunovic, A.; Eriksson Faelker, T.M.; et al. Mitochondrial dysfunction combined with high calcium load leads to impaired antioxidant defense underlying the selective loss of nigral dopaminergic neurons. J. Neurosci. Off. J. Soc. Neurosci. 2020, 40, 1975–1986. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volles, M.J.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- Scudamore, O.; Ciossek, T. Increased oxidative stress exacerbates alpha-synuclein aggregation in vivo. J. Neuropathol. Exp. Neurol. 2018, 77, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by alpha-synuclein. PLoS ONE 2017, 12, e0184040. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 14508–14520. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.M.; Zhang, F.; Zhou, H.; Kam, W.; Wilson, B.; Hong, J.S. Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ. Health Perspect. 2011, 119, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Sapru, M.K.; Yates, J.W.; Hogan, S.; Jiang, L.; Halter, J.; Bohn, M.C. Silencing of human alpha-synuclein in vitro and in rat brain using lentiviral-mediated RNAi. Exp. Neurol. 2006, 198, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, A.D.; Cannon, J.R.; Tapias, V.; Bai, Q.; Horowitz, M.P.; Shah, V.; El Ayadi, A.; Hastings, T.G.; Greenamyre, J.T.; Burton, E.A. shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J. Clin. Investig. 2015, 125, 2721–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, A.L.; Mak, S.K.; Henderson, J.M.; Bumcrot, D.; Farrer, M.J.; di Monte, D.A. Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS ONE 2010, 5, e12122. [Google Scholar] [CrossRef]
- Lewis, J.; Melrose, H.; Bumcrot, D.; Hope, A.; Zehr, C.; Lincoln, S.; Braithwaite, A.; He, Z.; Ogholikhan, S.; Hinkle, K.; et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener. 2008, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. Alpha-synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. Therapeutic antisense oligonucleotides for movement disorders. Med. Res. Rev. 2021, 41, 2656–2688. [Google Scholar] [CrossRef]
- Alarcon-Aris, D.; Recasens, A.; Galofre, M.; Carballo-Carbajal, I.; Zacchi, N.; Ruiz-Bronchal, E.; Pavia-Collado, R.; Chica, R.; Ferres-Coy, A.; Santos, M.; et al. Selective alpha-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: Potential therapy for Parkinson’s disease. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 550–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Targeting huntingtin expression in patients with Huntington’s disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef]
- Pavia-Collado, R.; Coppola-Segovia, V.; Miquel-Rio, L.; Alarcon-Aris, D.; Rodriguez-Aller, R.; Torres-Lopez, M.; Paz, V.; Ruiz-Bronchal, E.; Campa, L.; Artigas, F.; et al. Intracerebral administration of a ligand-ASO conjugate selectively reduces alpha-synuclein accumulation in monoamine neurons of double mutant human A30P*A53T*alpha-synuclein transgenic mice. Int. J. Mol. Sci. 2021, 22, 2939. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Choong, C.J.; Nakamori, M.; Hayakawa, H.; Nishiyama, K.; Kasahara, Y.; Baba, K.; Nagata, T.; Yokota, T.; Tsuda, H.; et al. Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting alpha-synuclein as a novel therapy for Parkinson’s disease. Sci. Rep. 2019, 9, 7567. [Google Scholar] [CrossRef]
- Hayashita-Kinoh, H.; Yamada, M.; Yokota, T.; Mizuno, Y.; Mochizuki, H. Down-regulation of alpha-synuclein expression can rescue dopaminergic cells from cell death in the substantia nigra of Parkinson’s disease rat model. Biochem. Biophys. Res. Commun. 2006, 341, 1088–1095. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhao, M.; Yan, R.; Liu, J.; Maddila, S.; Junn, E.; Mouradian, M.M. MicroRNA-7 protects against neurodegeneration induced by alpha-synuclein preformed fibrils in the mouse brain. Neurother. J. Am. Soc. Exp. Neurother. 2021. [Google Scholar] [CrossRef]
- Groiss, S.J.; Wojtecki, L.; Sudmeyer, M.; Schnitzler, A. Deep brain stimulation in Parkinson’s disease. Adv. Neurol. Disord. 2009, 2, 20–28. [Google Scholar] [CrossRef] [Green Version]
- Spencer, B.; Trinh, I.; Rockenstein, E.; Mante, M.; Florio, J.; Adame, A.; El-Agnaf, O.M.A.; Kim, C.; Masliah, E.; Rissman, R.A. Systemic peptide mediated delivery of an siRNA targeting alpha-syn in the CNS ameliorates the neurodegenerative process in a transgenic model of Lewy body disease. Neurobiol. Dis. 2019, 127, 163–177. [Google Scholar] [CrossRef]
- Bottger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef]
- Cooper, J.M.; Wiklander, P.B.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1476–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huey, R.; Hawthorne, S.; McCarron, P. The potential use of rabies virus glycoprotein-derived peptides to facilitate drug delivery into the central nervous system: A mini review. J. Drug Target. 2017, 25, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Schlich, M.; Longhena, F.; Faustini, G.; O’Driscoll, C.M.; Sinico, C.; Fadda, A.M.; Bellucci, A.; Lai, F. Anionic liposomes for small interfering ribonucleic acid (siRNA) delivery to primary neuronal cells: Evaluation of alpha-synuclein knockdown efficacy. Nano Res. 2017, 10, 3496–3508. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef] [Green Version]
- Xhima, K.; Nabbouh, F.; Hynynen, K.; Aubert, I.; Tandon, A. Noninvasive delivery of an alpha-synuclein gene silencing vector with magnetic resonance-guided focused ultrasound. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 1567–1579. [Google Scholar] [CrossRef]
- Meng, Y.; Pople, C.B.; Lea-Banks, H.; Abrahao, A.; Davidson, B.; Suppiah, S.; Vecchio, L.M.; Samuel, N.; Mahmud, F.; Hynynen, K.; et al. Safety and efficacy of focused ultrasound induced blood-brain barrier opening, an integrative review of animal and human studies. J. Control Release Off. J. Control Release Soc. 2019, 309, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.; Parikh, A.; Joshi, P.; Upadhyay, U.M.; Chotai, N.P. Intranasal drug delivery system—A glimpse to become maestro. J. Appl. Pharm. Sci. 2011, 1, 34–44. [Google Scholar]
- Erdo, F.; Bors, L.A.; Farkas, D.; Bajza, A.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Liu, X. Intranasal delivery: Circumventing the iron curtain to treat neurological disorders. Expert Opin. Drug Deliv. 2015, 12, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Volc, D.; Poewe, W.; Kutzelnigg, A.; Luhrs, P.; Thun-Hohenstein, C.; Schneeberger, A.; Galabova, G.; Majbour, N.; Vaikath, N.; El-Agnaf, O.; et al. Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol. 2020, 19, 591–600. [Google Scholar] [CrossRef]
- Affiris. Pipeline—Identifying Involved Proteins: Alpha Synuclein, mutHTT, PCSK9. Available online: https://affiris.com/pipeline/#clinical-trials (accessed on 18 November 2021).
- Schenk, D.B.; Koller, M.; Ness, D.K.; Griffith, S.G.; Grundman, M.; Zago, W.; Soto, J.; Atiee, G.; Ostrowitzki, S.; Kinney, G.G. First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Immunologic treatment of Parkinson’s disease. Immunotherapy 2018, 10, 81–84. [Google Scholar] [CrossRef]
- Brys, M.; Fanning, L.; Hung, S.; Ellenbogen, A.; Penner, N.; Yang, M.; Welch, M.; Koenig, E.; David, E.; Fox, T.; et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Adame, A.; Alford, M.; Crews, L.; Hashimoto, M.; Seubert, P.; Lee, M.; Goldstein, J.; Chilcote, T.; et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 2005, 46, 857–868. [Google Scholar] [CrossRef] [Green Version]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef] [Green Version]
- Clinicaltrials.gov. Study Assessing Tolerability and Safety of AFFITOPE® PD03A in Patients with Early Parkinson’s Disease. 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02267434 (accessed on 18 November 2021).
- AFFiRiS, AG. AFFiRiS Announces Top Line Results of First-in-Human Clinical Study Using AFFITOPE® PD03A, Confirming Immunogenicity and Safety Profile in Parkinson’s Disease. Available online: https://www.prnewswire.com/news-releases/affiris-announces-top-line-results-of-first-in-human-clinical-study-using-affitope-pd03a-confirming-immunogenicity-and-safety-profile-in-parkinsons-disease-patients-627025511.html (accessed on 18 November 2021).
- Chen, Z.; Yang, Y.; Yang, X.; Zhou, C.; Li, F.; Lei, P.; Zhong, L.; Jin, X.; Peng, G. Immune effects of optimized DNA vaccine and protective effects in a MPTP model of Parkinson’s disease. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2013, 34, 1559–1570. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Mante, M.; Crews, L.; Spencer, B.; Adame, A.; Patrick, C.; Trejo, M.; Ubhi, K.; Rohn, T.T.; et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE 2011, 6, e19338. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.J.; Lee, H.J.; Rockenstein, E.; Ho, D.H.; Park, E.B.; Yang, N.Y.; Desplats, P.; Masliah, E.; Lee, S.J. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 13454–13469. [Google Scholar] [CrossRef]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. A Study to Evaluate the Efficacy of Prasinezumab (RO7046015/PRX002) in Participants With Early Parkinson’s Disease (PASADENA). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03100149 (accessed on 18 November 2021).
- Schofield, D.J.; Irving, L.; Calo, L.; Bogstedt, A.; Rees, G.; Nuccitelli, A.; Narwal, R.; Petrone, M.; Roberts, J.; Brown, L.; et al. Preclinical development of a high affinity alpha-synuclein antibody, MEDI1341, that can enter the brain, sequester extracellular alpha-synuclein and attenuate alpha-synuclein spreading in vivo. Neurobiol. Dis. 2019, 132, 104582. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Single Ascending Dose Study of MEDI1341 in Healthy Volunteers. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03272165 (accessed on 18 November 2021).
- Clinicaltrials.gov. Multiple Ascending Dose Study of MEDI1341 in Patients with Parkinson’s Disease. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04449484 (accessed on 18 November 2021).
- Astrazenca.com. AstraZeneca and Takeda Establish Collaboration to Develop and Commercialise MEDI1341 for Parkinson’s Disease. Available online: https://www.astrazeneca.com/media-centre/press-releases/2017/astrazeneca-and-takeda-establish-collaboration-to-develop-and-commercialise-medi1341-for-parkinsons-disease-25082017.html# (accessed on 18 November 2021).
- Shahaduzzaman, M.; Nash, K.; Hudson, C.; Sharif, M.; Grimmig, B.; Lin, X.; Bai, G.; Liu, H.; Ugen, K.E.; Cao, C.; et al. Anti-human alpha-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-alpha-synuclein rat model of Parkinson’s disease. PLoS ONE 2015, 10, e0116841. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived alpha-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Evaluating the Efficacy, Safety, Pharmacokinetics, and Pharmacodynamics of BIIB054 in Participants with Parkinson’s Disease (SPARK). 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03318523 (accessed on 18 November 2021).
- Lindstrom, V.; Fagerqvist, T.; Nordstrom, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting alpha-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] alpha-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. A Study to Evaluate the Safety and Tolerability of ABBV-0805 in Patients with Parkinson’s Disease. 2020. Available online: https://clinicaltrials.gov/ct2/show/record/NCT04127695 (accessed on 18 November 2021).
- Kumar, S.T.; Jagannath, S.; Francois, C.; Vanderstichele, H.; Stoops, E.; Lashuel, H.A. How specific are the conformation-specific alpha-synuclein antibodies? Characterization and validation of 16 alpha-synuclein conformation-specific antibodies using well-characterized preparations of alpha-synuclein monomers, fibrils and oligomers with distinct structures and morphology. Neurobiol. Dis. 2020, 146, 105086. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.; Overk, C.; Rockenstein, E.; Mante, M.; Florio, J.; Adame, A.; Vaikath, N.; Majbour, N.; Lee, S.J.; Kim, C.; et al. Differential effects of immunotherapy with antibodies targeting alpha-synuclein oligomers and fibrils in a transgenic model of synucleinopathy. Neurobiol. Dis. 2017, 104, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, T.J.; Redmond, D.E., Jr.; Steece-Collier, K.; Lipton, J.W.; Manfredsson, F.P. Is alpha-synuclein loss-of-function a contributor to Parkinsonian pathology? Evidence from non-human primates. Front. Neurosci. 2016, 10, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbatyuk, O.S.; Li, S.; Nash, K.; Gorbatyuk, M.; Lewin, A.S.; Sullivan, L.F.; Mandel, R.J.; Chen, W.; Meyers, C.; Manfredsson, F.P.; et al. In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1450–1457. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Manfredsson, F.P. Loss of functional alpha-synuclein: A toxic event in Parkinson’s disease? J. Parkinson’s Dis. 2012, 2, 249–267. [Google Scholar] [CrossRef] [Green Version]
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Zharikov, A.; Bai, Q.; de Miranda, B.R.; van Laar, A.; Greenamyre, J.T.; Burton, E.A. Long-term RNAi knockdown of alpha-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol. Dis. 2019, 125, 146–153. [Google Scholar] [CrossRef]
- Connor-Robson, N.; Peters, O.M.; Millership, S.; Ninkina, N.; Buchman, V.L. Combinational losses of synucleins reveal their differential requirements for compensating age-dependent alterations in motor behavior and dopamine metabolism. Neurobiol. Aging 2016, 46, 107–112. [Google Scholar] [CrossRef] [Green Version]
- Ninkina, N.; Tarasova, T.V.; Chaprov, K.D.; Roman, A.Y.; Kukharsky, M.S.; Kolik, L.G.; Ovchinnikov, R.; Ustyugov, A.A.; Durnev, A.D.; Buchman, V.L. Alterations in the nigrostriatal system following conditional inactivation of alpha-synuclein in neurons of adult and aging mice. Neurobiol. Aging 2020, 91, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Parkinson’s, UK. The Prevalence and Incidence of Parkinson’s in the UK; Parkinson’s UK: London, UK, 2017. [Google Scholar]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Bockmann, A.; Meier, B.H.; et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [Green Version]
- Peng, C.; Gathagan, R.J.; Lee, V.M. Distinct alpha-Synuclein strains and implications for heterogeneity among alpha-Synucleinopathies. Neurobiol. Dis. 2018, 109, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.D.; Choi, M.L.; Ryten, M.; Hopkins, L.; Drews, A.; Botia, J.A.; Iljina, M.; Rodrigues, M.; Gagliano, S.A.; Gandhi, S.; et al. Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol. 2019, 137, 103–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal models of alpha-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef] [PubMed]
- Fayyad, M.; Salim, S.; Majbour, N.; Erskine, D.; Stoops, E.; Mollenhauer, B.; El-Agnaf, O.M.A. Parkinson’s disease biomarkers based on alpha-synuclein. J. Neurochem. 2019, 150, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; Wang, L.; Liu, W.; Zhu, G.; Chen, Y.; Zhang, J. Biomarkers and the role of alpha-synuclein in Parkinson’s disease. Front. Aging Neurosci 2021, 13, 645996. [Google Scholar] [CrossRef]
- Xu, M.M.; Ryan, P.; Rudrawar, S.; Quinn, R.J.; Zhang, H.Y.; Mellick, G.D. Advances in the development of imaging probes and aggregation inhibitors for alpha-synuclein. Acta Pharm. Sin. 2020, 41, 483–498. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
|
Author/Clinicaltrial.gov Identifier | Compound | Study Type | Status/Results |
|---|---|---|---|
| Active Immunization | |||
| NCT02267434 | PD03A | Phase Ib clinical trial in an early PD cohort (n = 36) | No serious adverse side effects, acceptable immune response against the vaccine, cross-reactivity against alpha-synuclein-targeted epitope |
| Volc et al., 2020 [62] | PD01A | Phase Ib clinical trial in an early PD cohort (n = 32) | PD01A antibodies were observed in CSF, demonstrating successful target engagement, acceptable levels of tolerability and safety |
| Affiris. 2021 [63] | PD01A | Phase II clinical trial (cohort type not yet specified) | Intention expressed |
| Passive Immunization | |||
| Schenk et al., 2017 [64] | PRX002 | Phase Ia clinical trial (n = 40) | Acceptable safety and tolerability; 95.5% reduction in serum alpha-synuclein |
| Jankovic et al., 2018 [65] | PRX002 | Phase Ib clinical trial in a mild to moderate PD cohort (n = 80) | Acceptable safety and tolerability; 95.5% reduction in serum alpha-synuclein and BBB penetration, dose-dependent rises of PRX002 measurements of CSF. |
| NCT03100149 | PRX002 | Phase II clinical trial in an early PD cohort (n = 316) | Active |
| NCT03272165 | MEDI1341 | Phase Ia clinical trial (n = 49) | Completed, awaiting results |
| NCT04449484 | MEDI1341 | Phase Ib clinical trial in a mild to moderate PD cohort (n = 36) | Recruiting |
| NCT04127695 | ABBV-0805 (formerly BAN0805) | Phase Ia clinical trial (n = 0) | Withdrawn for unspecified strategic reasons |
| Brys et al., 2019 [66] | BIIB054 | Phase Ib clinical trial, including a healthy (n = 48) and early PD cohort (n = 18) | Acceptable safety and tolerability; drug measured in the CSF (0.2% compared to plasma concentration) |
| NCT03318523 | BIIB054 | Phase II clinical trial in an early PD cohort (n = 357) | Active |
| Models | Positives | Negatives |
|---|---|---|
| Transgenic rodent model (e.g., Thy1-hA30P-alpha-synuclein) |
|
|
| Viral-vector delivery rodent model (e.g., AAV-hA53T) |
|
|
| PFF injection rodent model |
|
|
| WT non-human primate model |
|
|
| Viral-delivery non-human primate model (e.g., AAV1/2-hA53T) |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upcott, M.; Chaprov, K.D.; Buchman, V.L. Toward a Disease-Modifying Therapy of Alpha-Synucleinopathies: New Molecules and New Approaches Came into the Limelight. Molecules 2021, 26, 7351. https://doi.org/10.3390/molecules26237351
Upcott M, Chaprov KD, Buchman VL. Toward a Disease-Modifying Therapy of Alpha-Synucleinopathies: New Molecules and New Approaches Came into the Limelight. Molecules. 2021; 26(23):7351. https://doi.org/10.3390/molecules26237351
Chicago/Turabian StyleUpcott, Matthew, Kirill D. Chaprov, and Vladimir L. Buchman. 2021. "Toward a Disease-Modifying Therapy of Alpha-Synucleinopathies: New Molecules and New Approaches Came into the Limelight" Molecules 26, no. 23: 7351. https://doi.org/10.3390/molecules26237351
APA StyleUpcott, M., Chaprov, K. D., & Buchman, V. L. (2021). Toward a Disease-Modifying Therapy of Alpha-Synucleinopathies: New Molecules and New Approaches Came into the Limelight. Molecules, 26(23), 7351. https://doi.org/10.3390/molecules26237351