
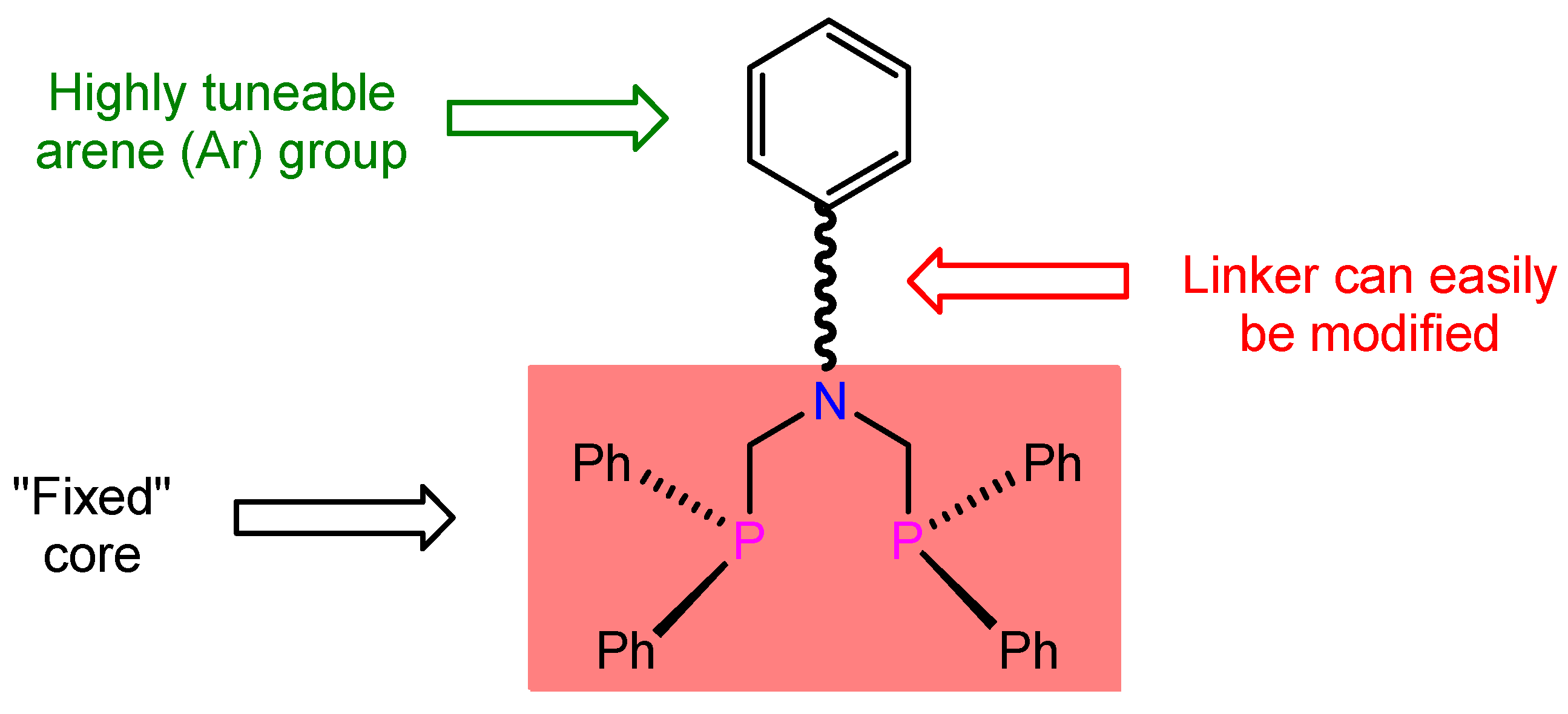
Low-Dimensional Architectures in Isomeric cis-PtCl2{Ph2PCH2N(Ar)CH2PPh2} Complexes Using Regioselective-N(Aryl)-Group Manipulation


Abstract
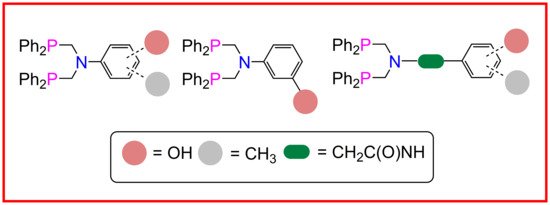
:
1. Introduction
2. Results and Discussion
2.1. Ligand Synthesis
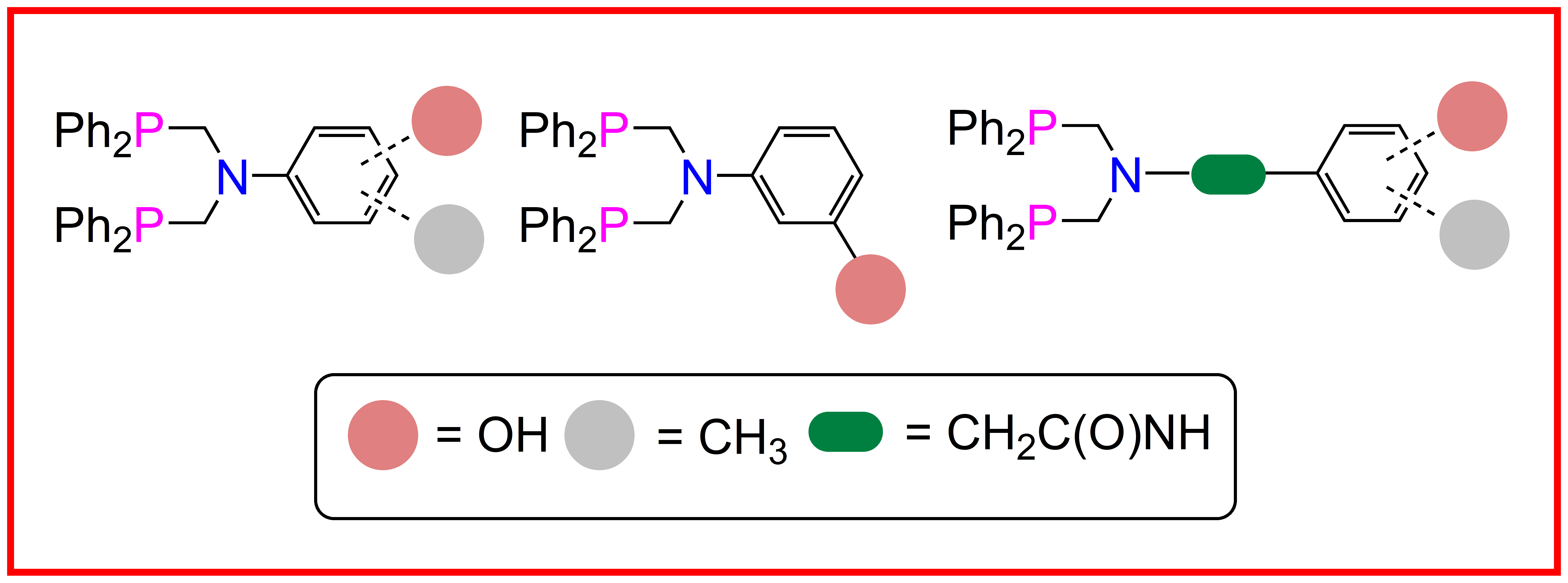
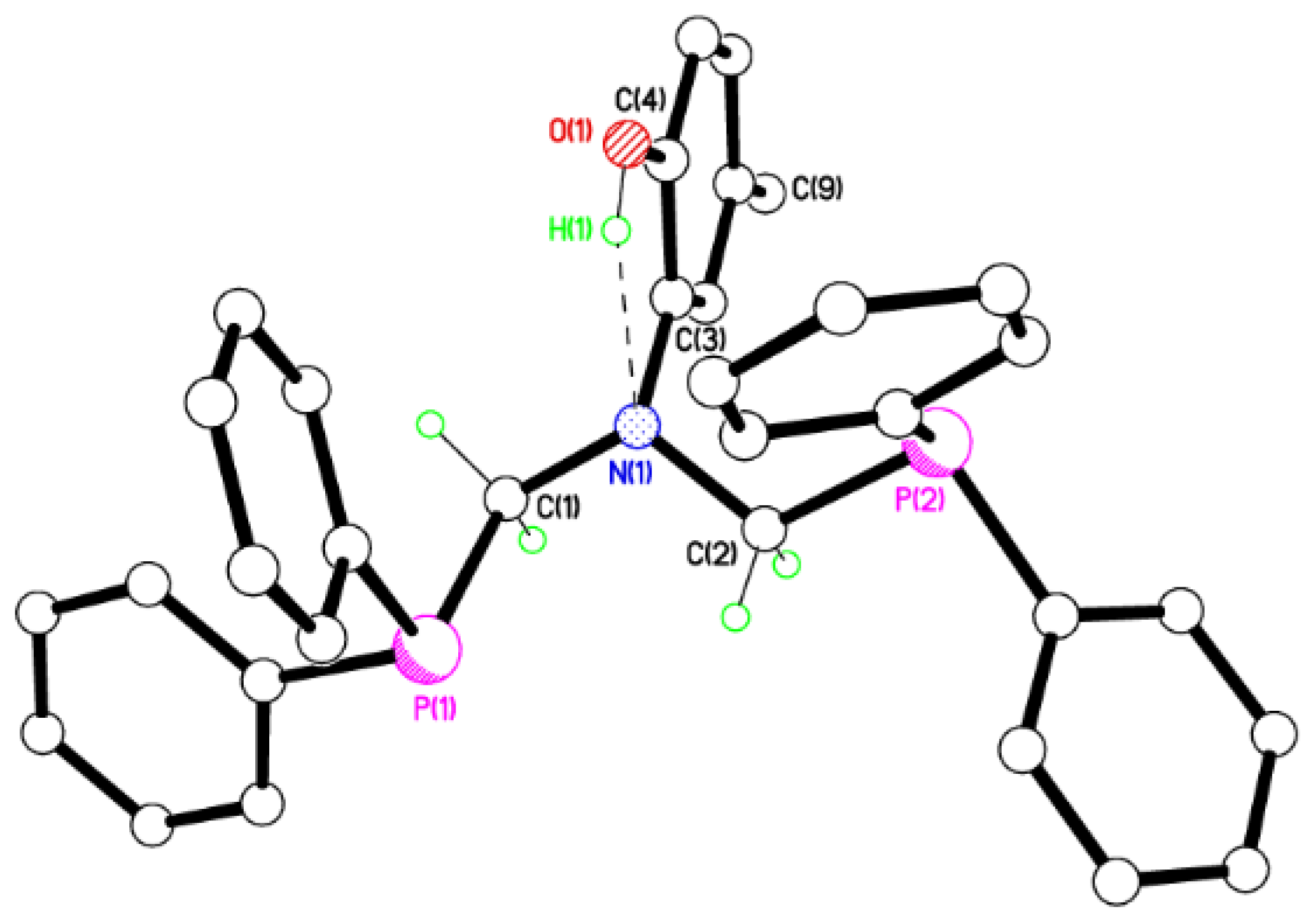
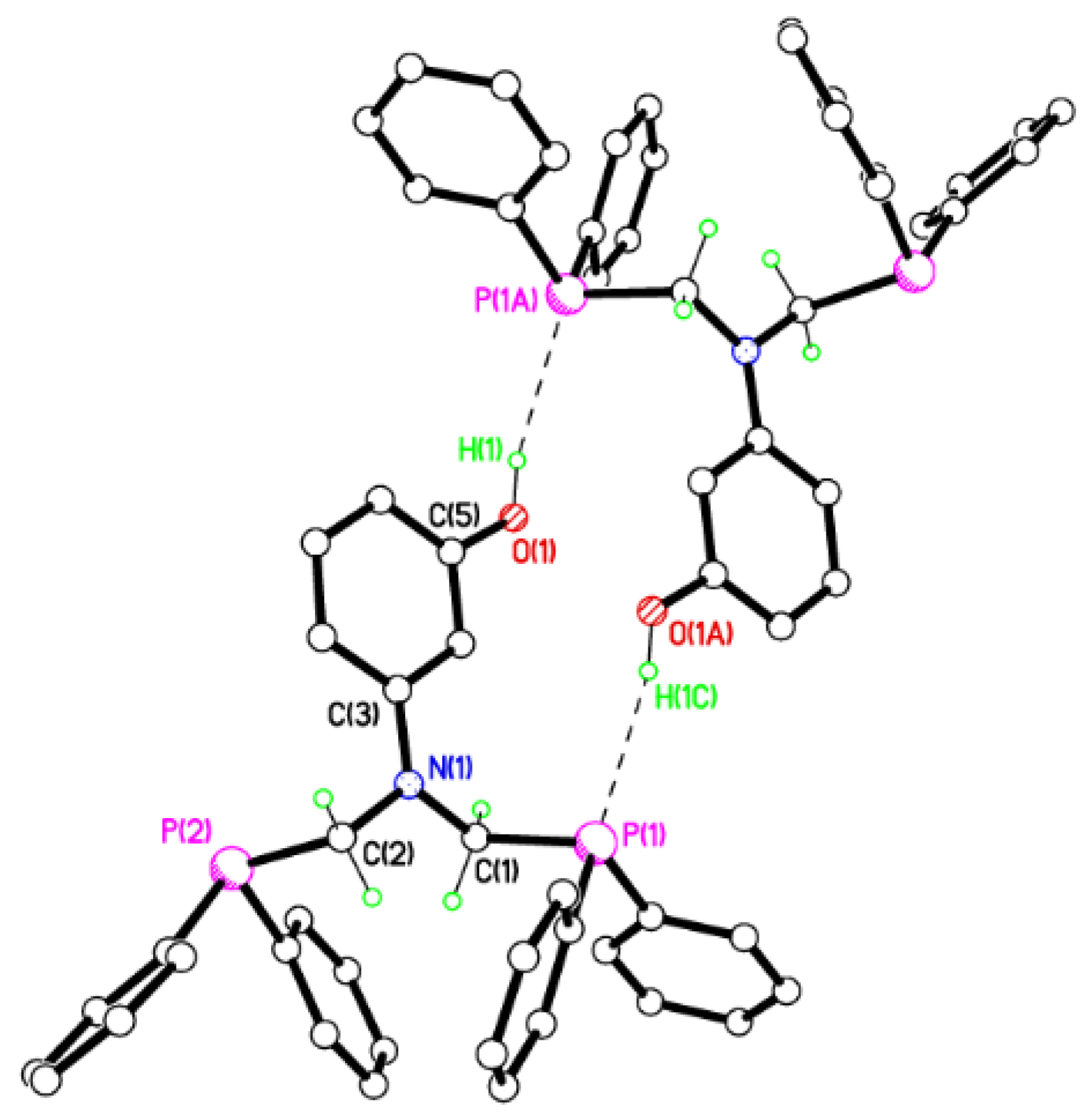
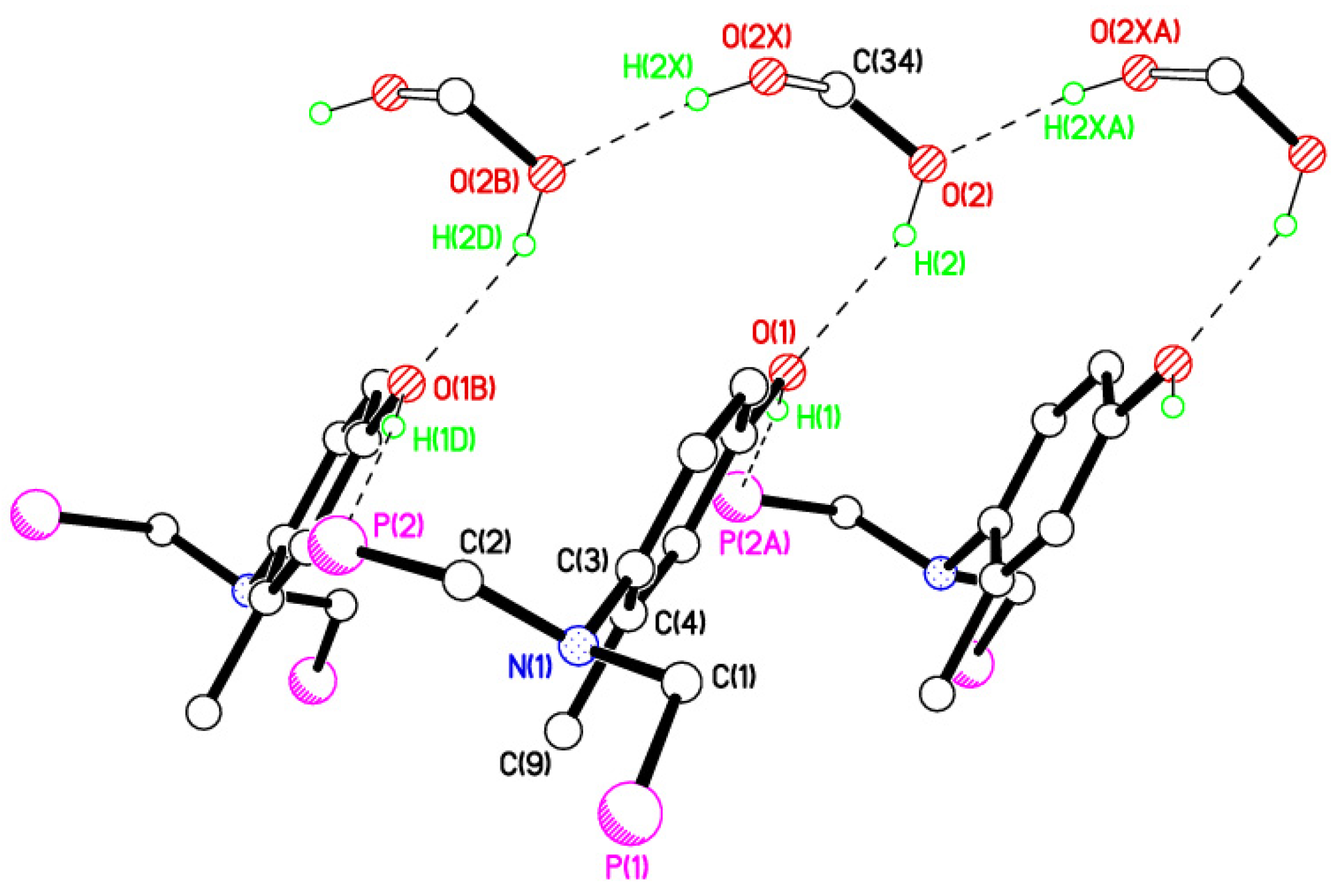
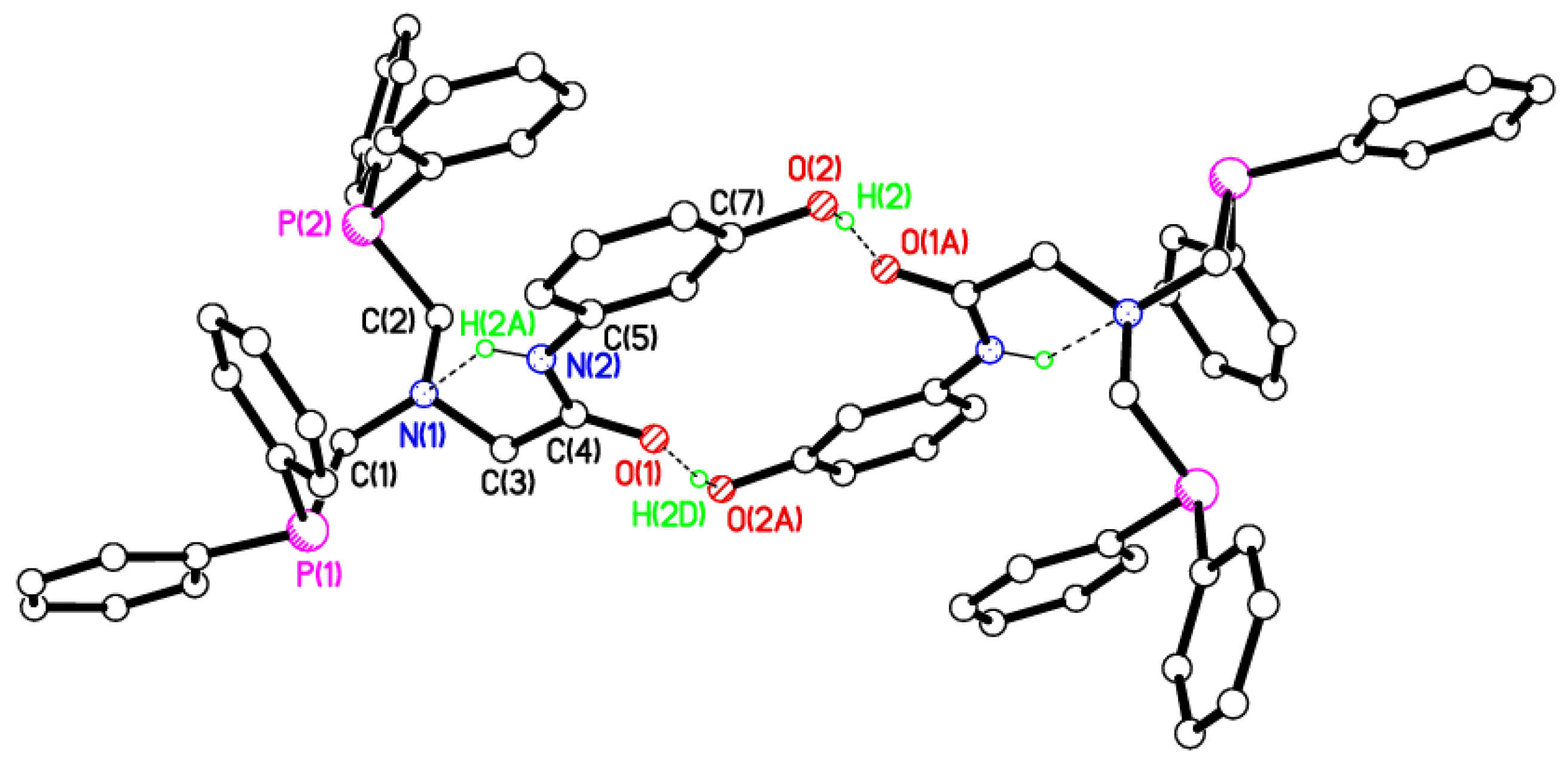
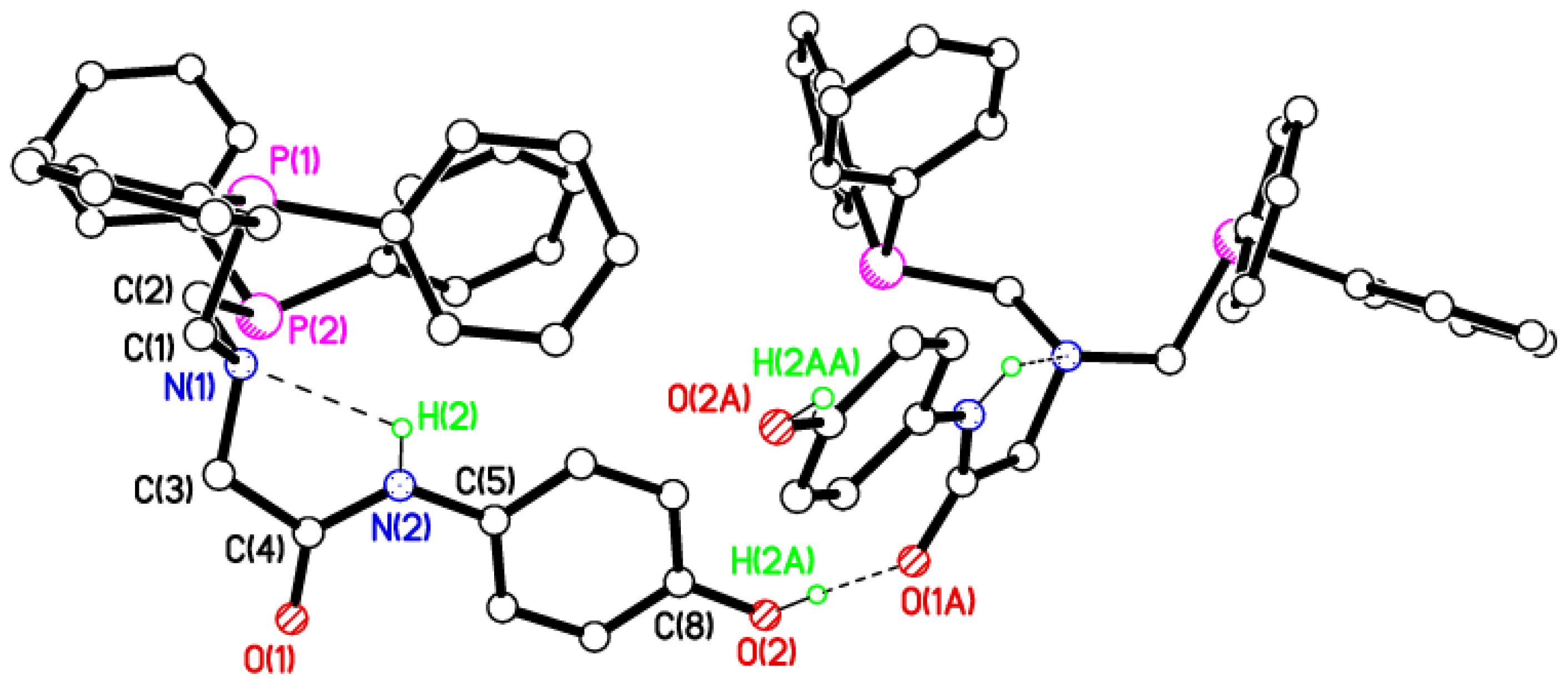
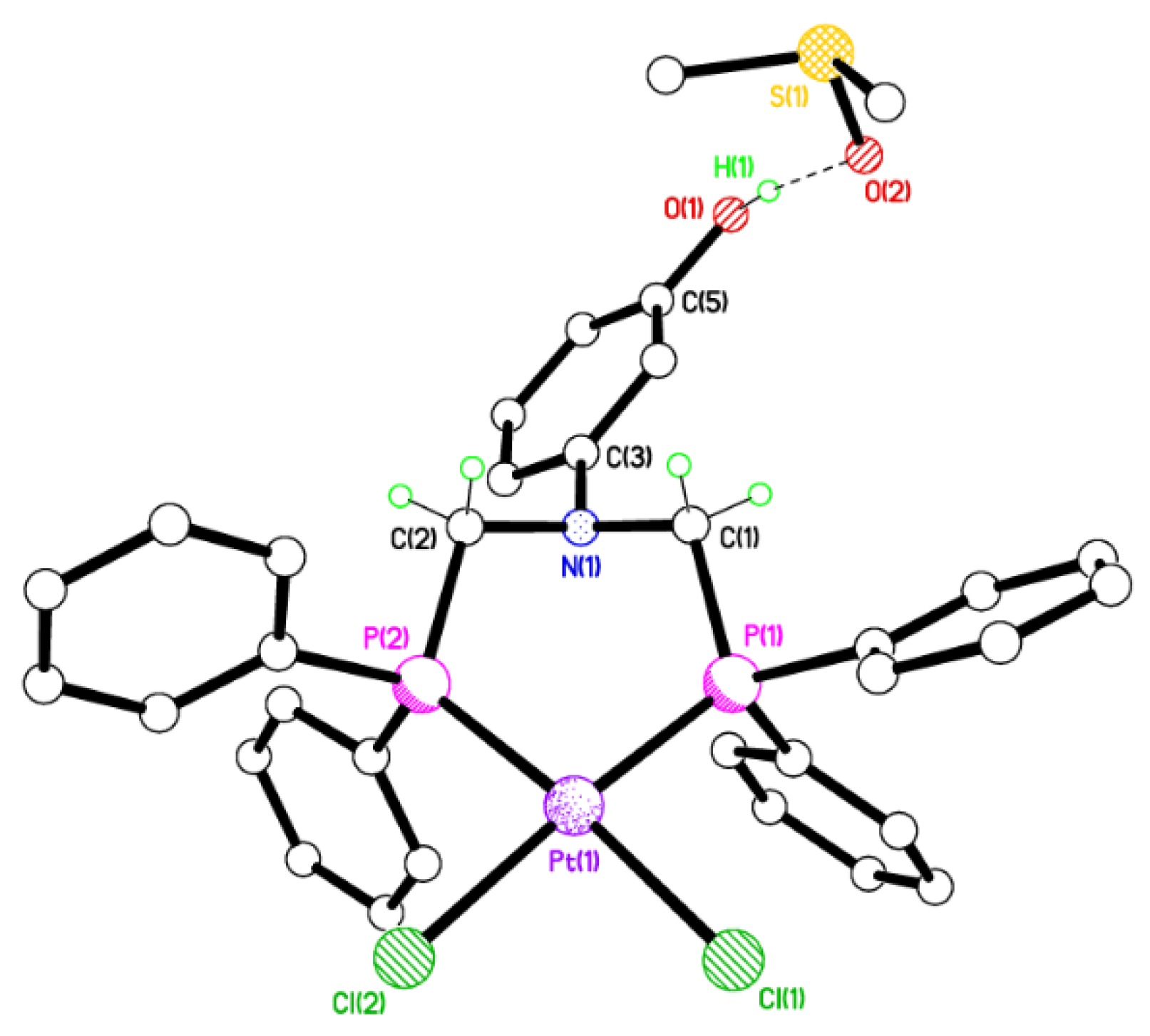
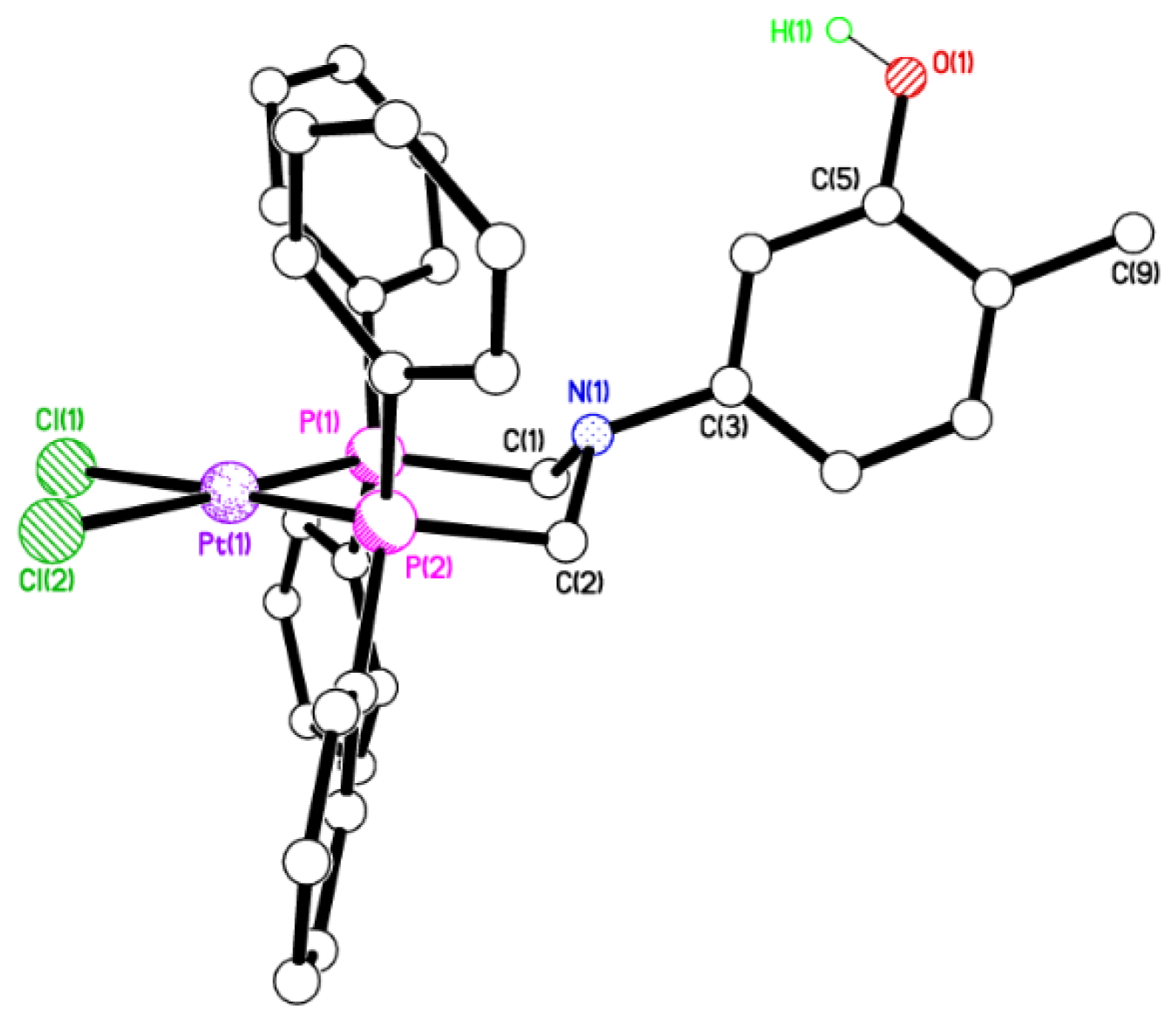
2.2. Single Crystal X-ray Studies of 1a, 1b∙CH3OH, 2f∙CH3OH, 2g, and 3
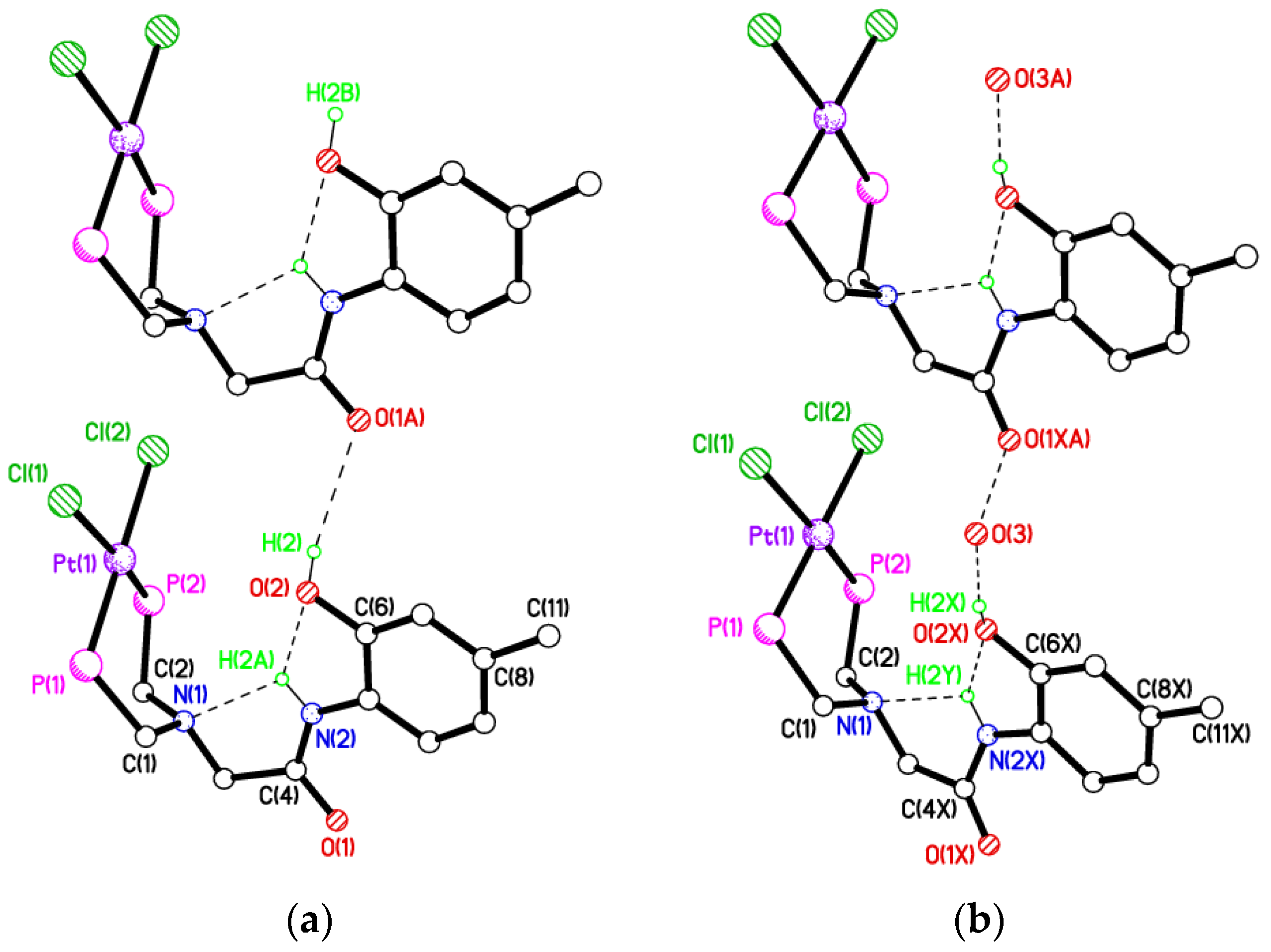
2.3. Secondary Interactions in 1a, 1b∙CH3OH, 2f∙CH3OH, 2g, and 3
2.4. Dichloroplatinum(II) Complexes of 1a–e, 2a–g, and 3
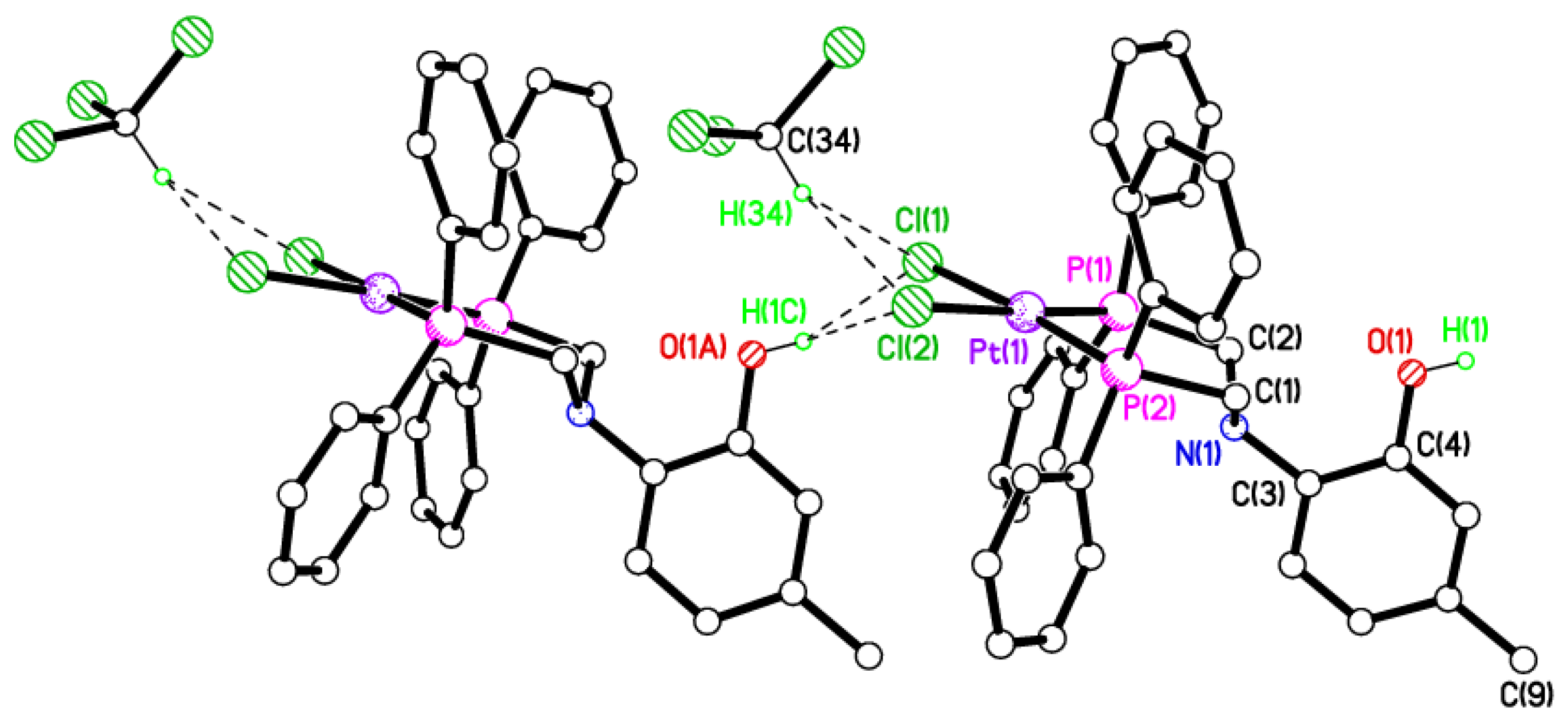
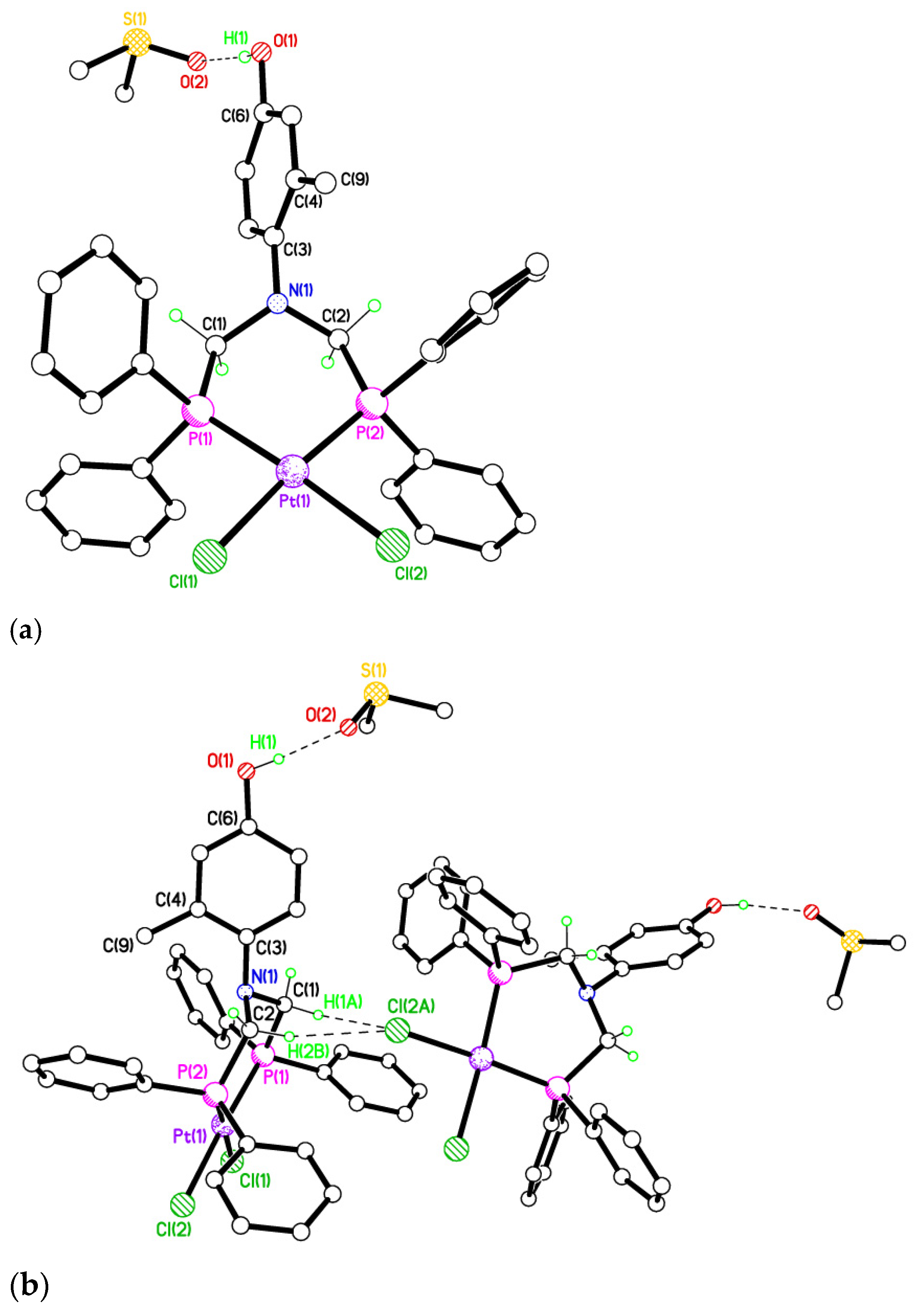
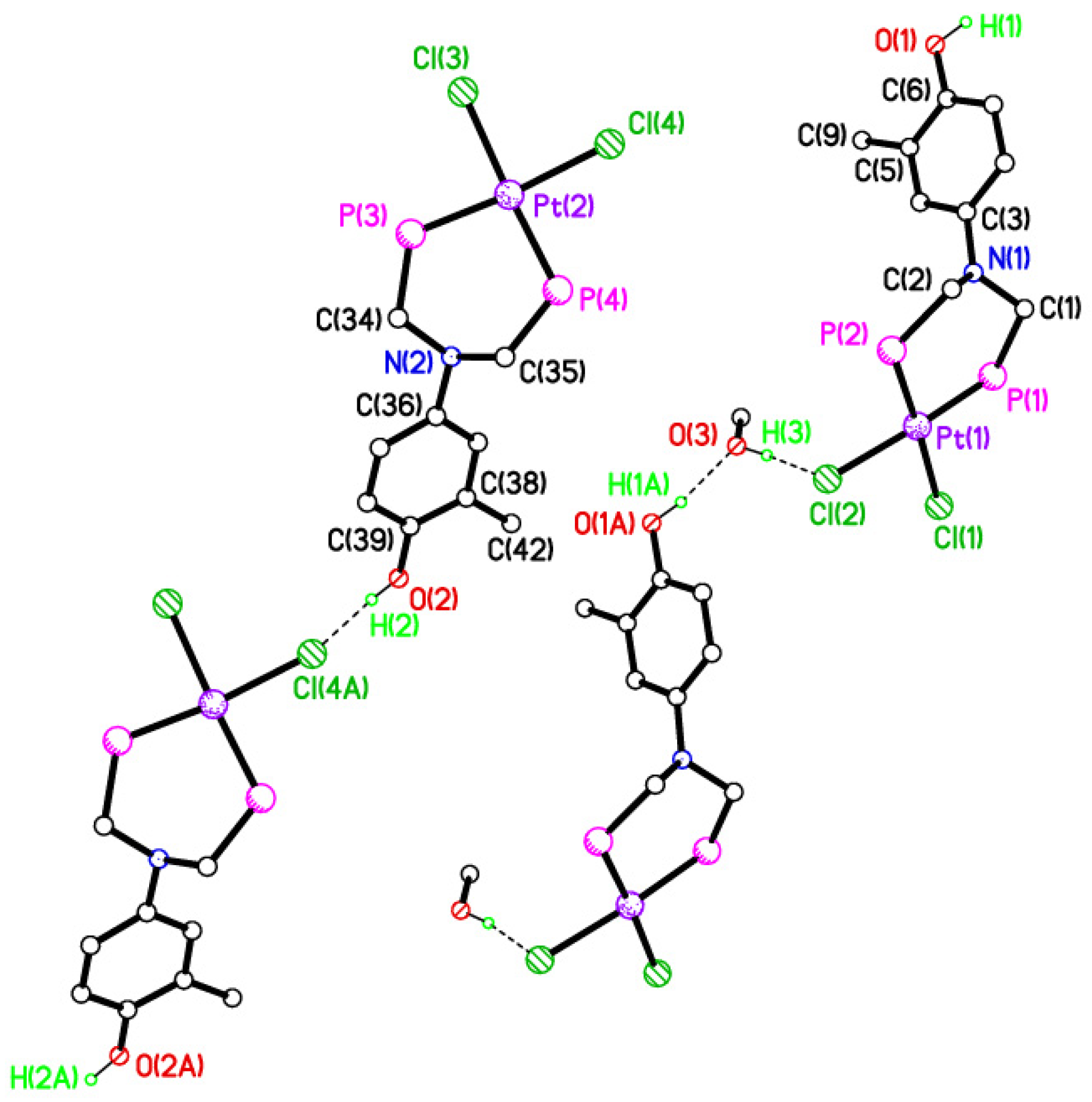
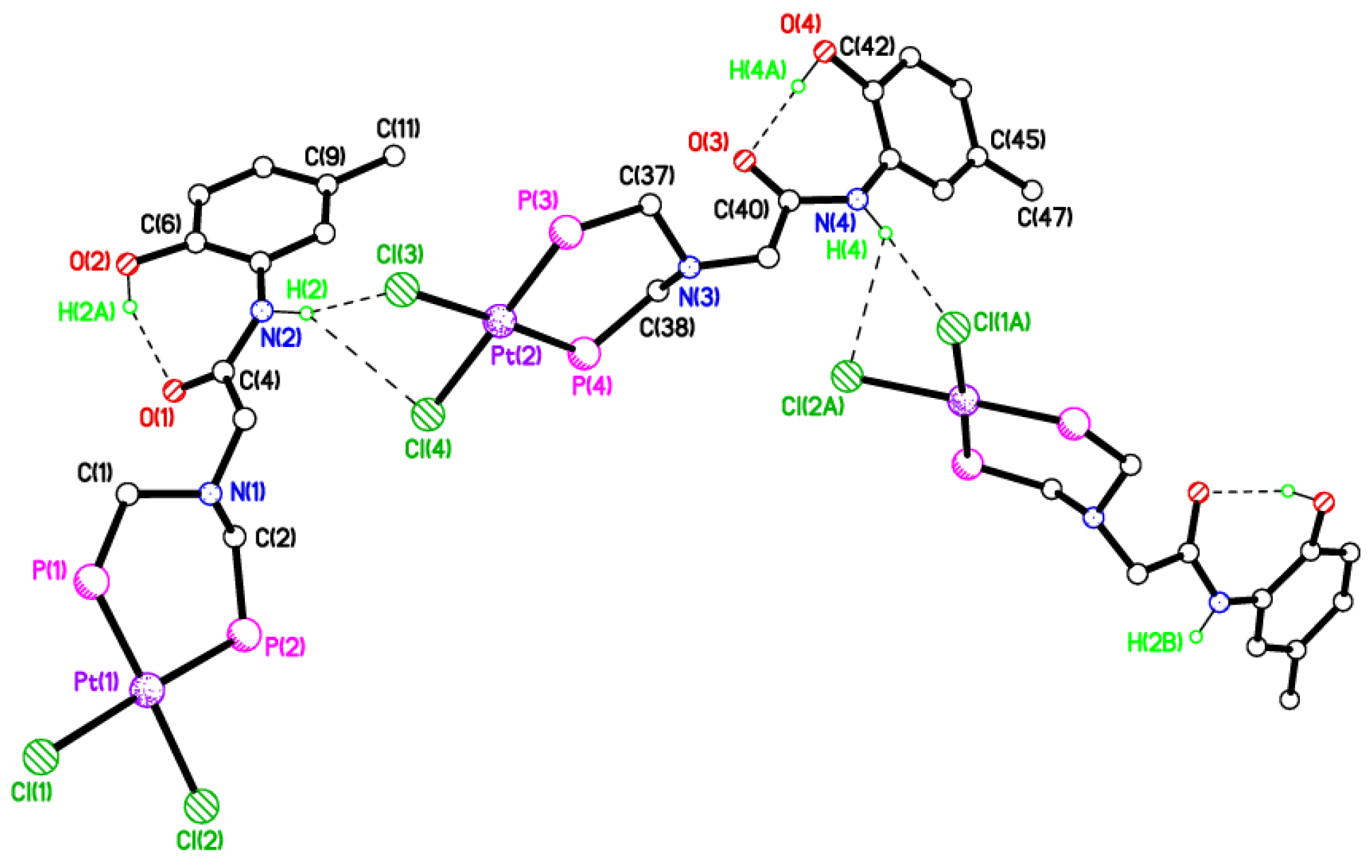
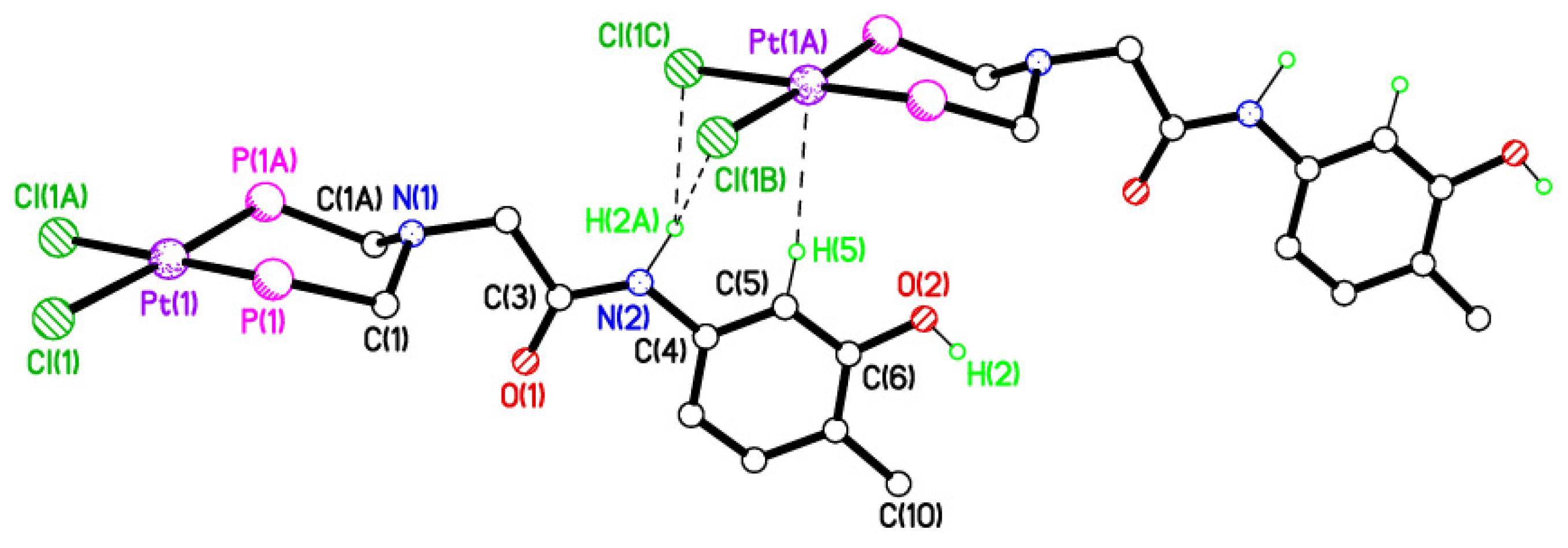
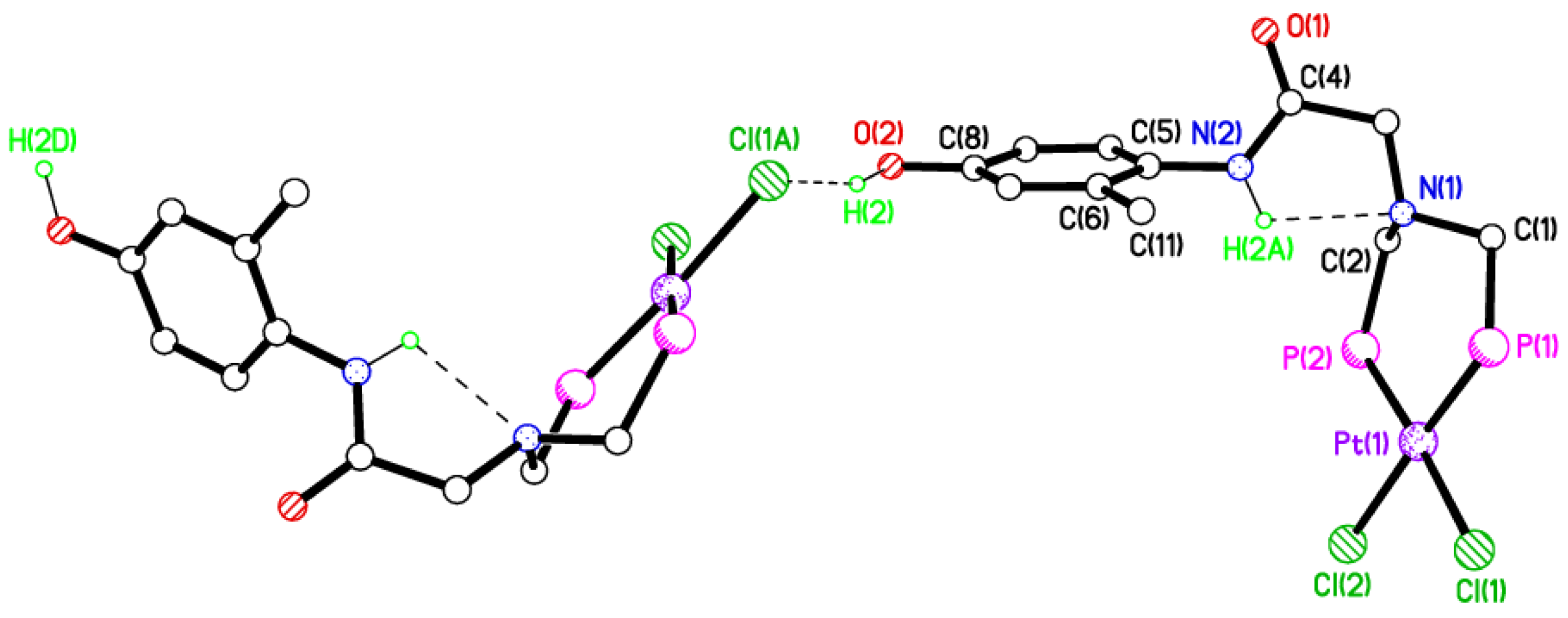
2.5. Single Crystal X-ray Studies of Complexes 4b∙(CH3)2SO, 4c∙CHCl3, 4d∙½Et2O, 4e∙½CHCl3∙½CH3OH, 5a∙½Et2O, 5b, 5c∙¼H2O, 5d∙Et2O, and 6∙(CH3)2SO
3. Conclusions
4. Materials and Methods
4.1. General Procedures
4.2. Instrumentation
4.3. Preparation of Ligands 1a–e, 2a–g, and 3
4.4. Preparation of cis-Dichloroplatinum(II) Phosphine Complexes 4a–e, 5a–g, and 6
4.5. Single Crystal X-ray Crystallography
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lehn, J.-M. Supramolecular Chemistry-Scope and Perspectives. Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Jongkind, L.J.; Caumes, X.; Hartendorp, A.P.T.; Reek, J.N.H. Ligand Template Strategies for Catalyst Encapsulation. Acc. Chem. Res. 2018, 51, 2115–2128. [Google Scholar] [CrossRef] [PubMed]
- James, S.L. Phosphines as building blocks in coordination-based self-assembly. Chem. Soc. Rev. 2009, 38, 1744–1758. [Google Scholar] [CrossRef]
- Breit, B. Supramolecular Approaches to Generate Libraries of Chelating Bidentate Ligands for Homogeneous Catalysis. Angew. Chem. Int. Ed. 2005, 44, 6816–6825. [Google Scholar] [CrossRef] [PubMed]
- Daubignard, J.; Detz, R.J.; de Bruin, B.; Reek, J.N.H. Phosphine Oxide Based Supramolecular Ligands in the Rhodium-Catalysed Asymmetric Hydrogenation. Organometallics 2019, 38, 3961–3969. [Google Scholar] [CrossRef]
- Koshti, V.S.; Sen, A.; Shinde, D.; Chikkali, S.H. Self-assembly of P-chiral supramolecular phosphines on rhodium and direct evidence for Rh-catalyst-substrate interactions. Dalton Trans. 2017, 46, 13966–13973. [Google Scholar] [CrossRef]
- Vasseur, A.; Membrat, R.; Palpacelli, D.; Giorgi, M.; Nuel, D.; Giordano, L.; Martinez, A. Synthesis of chiral supramolecular bisphosphinite palladcycles through hydrogen transfer-promoted self-assembly process. Chem. Commun. 2018, 54, 10132–10135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Nieto, C.; de Cózar, A.; Regulska, E.; Mullenix, J.B.; Rominger, F.; Hindenberg, P. Controlling the molecular arrangement of racemates through weak interactions: The synergy between p-interactions and halogen bonds. Chem. Commun. 2021, 57, 7366–7369. [Google Scholar] [CrossRef] [PubMed]
- Carreras, L.; Serrano-Torné, M.; van Leeuwen, P.W.N.M.; Vidal-Ferran, A. XBphos-Rh: A halogen-bond assembled supramolecular catalyst. Chem. Sci. 2018, 9, 3644–3648. [Google Scholar] [CrossRef] [Green Version]
- García-Márquez, A.; Frontera, A.; Roisnel, T.; Gramage-Doria, R. Ultrashort Hd+…Hd- intermolecular distance in a supramolecular system in the solid state. Chem. Commun. 2021, 57, 7112–7115. [Google Scholar] [CrossRef]
- Blann, K.; Bollmann, A.; Brown, G.M.; Dixon, J.T.; Elsegood, M.R.J.; Raw, C.R.; Smith, M.B.; Tenza, K.; Willemse, A.; Zweni, P. Ethylene oligomerisation chromium catalysts with unsymmetrical PCNP ligands. Dalton Trans. 2021, 50, 4345–4354. [Google Scholar] [CrossRef] [PubMed]
- De’Ath, P.; Elsegood, M.R.J.; Halliwell, C.A.G.; Smith, M.B. Mild intramolecular P-C(sp3) bond cleavage in bridging diphosphine complexes of RuII, RhIII, and IrIII. J. Organomet. Chem. 2021, 937, 121704. [Google Scholar] [CrossRef]
- Smith, M.B.; Dale, S.H.; Coles, S.J.; Gelbrich, T.; Hursthouse, M.B.; Light, M.E.; Horton, P.N. Hydrogen bonded supramolecular assemblies based on neutral square-planar palladium(II) complexes. CrystEngCommun 2007, 9, 165–175. [Google Scholar] [CrossRef]
- Smith, M.B.; Dale, S.H.; Coles, S.J.; Gelbrich, T.; Hursthouse, M.B.; Light, M.E. Isomeric dinuclear gold(I) complexes with highly functionalised ditertiary phosphines: Self-assembly of dimers, rings and 1-D polymeric chains. CrystEngCommun 2006, 8, 140–149. [Google Scholar] [CrossRef]
- Dann, S.E.; Durran, S.E.; Elsegood, M.R.J.; Smith, M.B.; Staniland, P.M.; Talib, S.; Dale, S.H. Supramolecular chemistry of half-sandwich organometallic building blocks based on RuCl2(p-cymene)Ph2PCH2Y. J. Organomet. Chem. 2006, 691, 4829–4842. [Google Scholar] [CrossRef]
- Durran, S.E.; Smith, M.B.; Slawin, A.M.Z.; Gelbrich, T.; Hursthouse, M.B.; Light, M.E. Synthesis and coordination studies of new aminoalcohol functionalised tertiary phosphines. Can. J. Chem. 2001, 79, 780–791. [Google Scholar] [CrossRef]
- Jiang, M.-S.; Tao, Y.-H.; Wang, Y.-W.; Lu, C.; Young, D.J.; Lang, J.-P.; Ren, Z.-G. Reversible Solid-State Phase Transitions between Au-P Complexes Accompanied by Switchable Fluorescence. Inorg. Chem. 2020, 59, 3072–3078. [Google Scholar] [CrossRef]
- Pandey, M.K.; Kunchur, H.S.; Mondal, D.; Radhakrishna, L.; Kote, B.S.; Balakrishna, M.S. Rare Au…H Interactions in Gold(I) Complexes of Bulky Phosphines Derived from 2,6-Dibenzhydryl-4-methylphenyl Core. Inorg. Chem. 2020, 59, 3642–3658. [Google Scholar] [CrossRef] [PubMed]
- Bálint, E.; Tajti, A.; Tripolszky, A.; Keglevich, G. Synthesis of platinum, palladium and rhodium complexes of α-aminophosphine ligands. Dalton Trans. 2018, 47, 4755–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-P.; Zhang, M.; Chen, X.-R.; Lu, C.; Young, D.J.; Ren, Z.-G.; Lang, J.-P. Cobalt(I) and Nickel(II) Complexes of a PNN Type Ligand as Photoenhanced Electrocatalysts for the Hydrogen Evolution Reaction. Inorg. Chem. 2020, 59, 1038–1045. [Google Scholar] [CrossRef]
- Hou, R.; Huang, T.-H.; Wang, X.-J.; Jiang, X.-F.; Ni, Q.-L.; Gui, L.-C.; Fan, Y.-J.; Tan, Y.-L. Synthesis, structural characterisation and luminescent properties of a series of Cu(I) complexes based on polyphosphine ligands. Dalton Trans. 2011, 40, 7551–7558. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-J.; Gui, L.-C.; Ni, Q.-L.; Liao, Y.-F.; Jiang, X.-F.; Tang, L.-H.; Zhang, Z.; Wu, Q. p-Stacking induced complexes with Z-shape motifs featuring a complimentary approach between electron-rich arene diamines and electron-deficient aromatic N-heterocycles. CrystEngComm 2008, 10, 1003–1010. [Google Scholar] [CrossRef]
- Au, R.H.W.; Jennings, M.C.; Puddephatt, R.J. Supramolecular Organoplatinum(IV) Chemistry: Sequential Introduction of Amide Hydrogen Bonding Groups. Organometallics 2009, 28, 3754–3762. [Google Scholar] [CrossRef]
- Coles, N.T.; Gasperini, D.; Provis-Evans, C.B.; Mahon, M.F.; Webster, R.L. Heterobimetallic Complexes of 1,1-Diphosphineamide Ligands. Organometallics 2021, 40, 148–155. [Google Scholar] [CrossRef]
- Navrátil, M.; Císařová, I.; Alemayehu, A.; Škoch, K.; Štěpnička, P. Synthesis and Structural Characterisation of an N-Phosphanyl Ferrocene Carboxamide and its Ruthenium, Rhodium and Palladium Complexes. ChemPlusChem 2020, 85, 1325–1338. [Google Scholar] [CrossRef]
- Navrátil, M.; Císařová, I.; Štěpnička, P. Intermolecular interactions in the crystal structures of chlorogold(I) complexes with N-phosphinoamide ligands. Inorg. Chim. Acta 2021, 516, 120138. [Google Scholar] [CrossRef]
- Pachisia, S.; Kishan, R.; Yadav, S.; Gupta, R. Half-Sandwich Ruthenium Complexes of Amide-Phosphine Based Ligands: H-Bonding Cavity Assisted Binding and Reduction of Nitro-substrates. Inorg. Chem. 2021, 60, 2009–2022. [Google Scholar] [CrossRef] [PubMed]
- Nasser, N.; Eisler, D.J.; Puddephatt, R.J. A chiral diphosphine as trans-chelate ligand and its relevance to catalysis. Chem. Commun. 2010, 46, 1953–1955. [Google Scholar] [CrossRef] [PubMed]
- Elsegood, M.R.J.; Lake, A.J.; Smith, M.B.; Weaver, G.W. Ditertiary phosphines bearing a -N-C-C(O)-N(H)- linker and their corresponding dichloroplatinum(II) complexes. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 540–544. [Google Scholar] [CrossRef]
- Hoyos, O.L.; Bermejo, M.R.; Fondo, M.; García-Deibe, A.; González, A.M.; Maneiro, M.; Pedrido, R. Mn(III) complexes with asymmetrical N2O3Schiff bases. The unusual crystal structure of [Mn(phenglydisal-3-Br,5-Cl)(dmso)] (H3phenglydisal = 3-aza-N-{2-[1-aza-2-(2-hydroxyphenyl)vinyl]phenyl}-4-(2-hydroxyphenyl)but-3-enamide), a mononuclear single-stranded helical manganese(III) complex. J. Chem. Soc. Dalton Trans. 2000, 3122–3127. [Google Scholar] [CrossRef]
- Bermejo, M.R.; González, A.M.; Fondo, M.; García-Deibe, A.; Maneiro, M.; Sanmartín, J.; Hoyos, O.L.; Watkinson, M. A direct route to obtain manganese(III) complexes with a new class of asymmetrical Schiff base ligands. New J. Chem. 2000, 24, 235–241. [Google Scholar] [CrossRef]
- Elsegood, M.R.J.; Smith, M.B.; Staniland, P.M. Neutral Molecular Pd6 Hexagons Using k3-P2O Terdentate Ligands. Inorg. Chem. 2006, 45, 6761–6770. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-Set Analysis of Hydrogen-Bond Patterns in Organic Crystals. Acta Crystallogr. 1990, B46, 256–262. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and Decoding Hydrogen-Bond Patterns of Organic Compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.-L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- Desiraju, G.; Steiner, T. The Weak Hydrogen Bond; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Klemps, C.; Payet, E.; Magna, L.; Saussine, L.; Le Goff, X.F.; Le Floch, P. PCNCP Ligands in the Chromium-Catalysed Oligomerisation of Ethylene: Tri-versus Tetramerization. Chem. Eur. J. 2009, 15, 8259–8268. [Google Scholar] [CrossRef]
- Walsh, A.P.; Laureanti, J.A.; Katipamula, S.; Chambers, G.M.; Priyadarshani, N.; Lense, S.; Bays, J.T.; Linehan, J.C.; Shaw, W.J. Evaluating the impacts of amino acids in the second and outer coordination spheres of Rh-bis(diphosphine) complexes for CO2 hydrogenation. Faraday Discuss. 2019, 215, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Hellman, H.; Bader, J.; Birkner, H.; Schumacher, O. Hydroxymethyl-phosphine, Hydroxymethyl-phosphoniumsalze und Chlormethyl-phosphoniumsalze. Justus Liebigs Ann. Chem. 1962, 659, 49–56. [Google Scholar] [CrossRef]
- McDermott, J.X.; White, J.F.; Whitesides, G.M. Thermal Decomposition of Bis(phosphine)platinum(II) Metallocycles. J. Am. Chem. Soc. 1976, 98, 6521–6528. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C-Struct. Chem. 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Sluis, P.v.d.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. 1990, A46, 194–201. [Google Scholar] [CrossRef]
- Bruker SMART Version 5.611; Bruker AXS Inc.: Fitchburg, WI, USA, 2001.
- Area-Detector Integration Software, APEX-II, Version V1; Bruker-Nonius: Madison, WI, USA, 2004.
- Denzo, Z.; Otwinowski, W. Processing of X-ray diffraction data in oscillation mode, Methods in Enzymology. In Macromolecular Crystallography; Carter, C.W., Jr., Sweet, R.M., Eds.; Academic Press: Cambridge, MA, USA, 1997; Volume 276, pp. 307–326. [Google Scholar]
- Hooft, R.W.W. COLLECT: Data Collection Software; Nonius B.V.: Delft, The Netherlands, 1998. [Google Scholar]
- SAINT Software for CCD Diffractometers; Bruker AXS Inc.: Madison, WI, USA, 2004.
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D.J. SADABS software. J. Appl. Cryst. 2015, 48, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXTL User Manual, Version 6.12; Bruker AXS Inc.: Madison, WI, USA, 2001. [Google Scholar]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Com-pound a | δ(P) b | δ(H) /OH (NH) | δ(H) /arom. H. | δ(H) /CH2 | δ(H) /CH2 d | δ(H) /CH3 | νOH (νNH) e | Microanalysis (CHN) |
|---|---|---|---|---|---|---|---|---|
| 1a (79) | −27.5 | 8.62 | 7.33–7.23, 6.76, 6.69–6.57 | 4.15 (2.4) | 2.10 | 3398 | Calc. for C33H31NOP2, C, 76.29; H, 6.01; N, 2.70 Found, C, 76.07; H, 6.13; N, 2.78 | |
| 1b (56) | −27.3 | 9.06 | 7.36–7.26, 7.15, 6.50, 6.44 | 3.96 (5.6) | 1.74 | 3282 | Calc. for C33H31NOP2.2MeOH, C, 72.03; H, 6.74; N, 2.40 Found, C, 72.45; H, 6.04; N, 2.58 | |
| 1c (97) | −27.5 | 8.77 | 7.44–7.22, 6.86, 6.54, 6.48 | 4.09 (3.4) | 2.12 | 3389 | Calc. for C33H31NOP2, C, 76.29; H, 6.01; N, 2.70 Found, C, 75.99; H, 6.00; N, 2.76 | |
| 1d (38) | −26.7 | 8.63 | 7.40–7.30, 6.55 | 4.02 (3.2) | 1.96 | 3432 | Calc. for C33H31NOP2, C, 76.29; H, 6.01; N, 2.70 Found, C, 75.53; H, 6.05; N, 2.74 | |
| 1e (96) | −26.4 | 9.06 | 7.49–7.33, 6.85, 6.50, 6.27 | 3.88 (3.6) | 2.08 | 3387 | Calc. for C33H31NOP2.MeOH, C, 74.03; H, 6.40; N, 2.54 Found, C, 74.81; H, 5.93; N, 2.61 | |
| 2a (81) | −26.0 | 8.15 | 7.77–7.19 | 5.06 | 3.62 (8.0) | 1.19 | - | - |
| 2b (89) | −26.0 | 7.83 | 7.60–7.21 | 5.07 | 3.69 (3.6) | 1.63 | - | - |
| 2c (88) | −26.5 c | 9.34 (8.17) | 7.71–7.19 | 5.27 | 3.61 (4.8) | 1.63 | - | - |
| 2d (65) | −27.1 | 9.05 (8.68) | 7.55–7.32, 6.95, 6.61, 6.41 | 3.69 | 3.81 (4.8) | 2.04 | 3047 (3228) | Calc. for C35H34N2O2P2, C, 72.91; H, 5.94; N, 4.86 Found, C, 72.72; H, 5.95; N, 4.88 |
| 2e (80) | −27.1 | 9.29 (9.08) | 7.46–7.35, 7.29, 6.86, 6.15 | 3.73 | 3.82 (4.4) | 2.08 | 3178 (3317) | Calc. for C35H34N2O2P2, C, 72.91; H, 5.94; N, 4.86 Found, C, 72.71; H, 5.94; N, 4.82 |
| 2f (70) | −27.5 | 9.31 (9.07) | 7.41–7.03, 6.94, 6.40, 6.30 | 3.69 | 3.77 (4.4) | 3163 (3283) | Calc. for C34H32N2O2P2, C, 72.59; H, 5.73; N, 4.98 Found, C, 72.10; H, 5.80; N, 4.95 | |
| 2g (85) | −26.8 | 9.09 (8.78) | 7.36–7.25, 6.83, 6.53 | 3.61 | 3.72 (4.4) | 3300 (3257) | Calc. for C34H32N2O2P2, C, 72.59; H, 5.73; N, 4.98 Found, C, 72.15; H, 5.72; N, 4.95 | |
| 3 (53) | −27.6 | 9.12 | 7.38–7.31, 6.92, 6.30, 6.13 | 3.85 | 3376 | Calc. for C32H29NOP2, C, 76.03; H, 5.78; N, 2.77 Found, C, 75.67; H, 5.71; N, 2.74 |
| Compound | 1a | 1b·CH3OH | 2f·CH3OH | 2g | 3 |
|---|---|---|---|---|---|
| Formula | C33H31NOP2 | C34H35NO2P2 | C35H36N2O3P2 | C34H32N2O2P2 | C32H29NOP2 |
| M | 519.53 | 551.57 | 594.60 | 562.55 | 505.50 |
| Crystal dimensions | 0.42 × 0.15 × 0.03 | 0.13 × 0.12 × 0.02 | 0.24 × 0.18 × 0.16 | 0.25 × 0.18 × 0.15 | 0.31 × 0.28 × 0.03 |
| Crystal morphology and colour | Plate, colourless | Block, colourless | Block, colourless | Block, colourless | Plate, colourless |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic | Triclinic |
| Space group | P21/n | P21/c | P | Ia | P |
| a/Å | 17.367(5) | 10.3050(3) | 12.6198(3) | 11.6234(10) | 10.5860(4) |
| b/Å | 8.522(2) | 32.8017(10) | 16.2027(4) | 21.7359(19) | 10.7397(4) |
| c/Å | 20.382(6) | 8.5189(2) | 17.8529(4) | 11.6340(10) | 13.4172(6) |
| α/° | 64.0678(10) | 73.1667(6) | |||
| β/° | 114.673(4) | 92.7318(16) | 76.7403(14) | 93.8717(14) | 80.4518(7) |
| γ/° | 75.5070(14) | 63.1422(6) | |||
| V/Å3 | 2741.2(13) | 2876.30(14) | 3148.15(13) | 2932.6(4) | 1301.45(9) |
| Z | 4 | 4 | 4 | 4 | 2 |
| λ/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Τ/Κ | 150(2) | 120(2) | 120(2) | 150(2) | 150(2) |
| Density (calcd.)/Mg/m3 | 1.259 | 1.274 | 1.255 | 1.274 | 1.290 |
| μ/mm−1 | 0.185 | 0.183 | 0.176 | 0.182 | 0.193 |
| θ range/° | 2.02–26.60 | 3.03–27.53 | 3.24–25.00 | 1.87–28.82 | 1.59–30.62 |
| Measured reflections | 23,525 | 27,247 | 61,330 | 12,577 | 15,576 |
| Independent reflections | 5708 | 6545 | 11,047 | 6586 | 7814 |
| Observed reflections (F2 > 2σ(F2)) | 3115 | 5019 | 7559 | 5293 | 6116 |
| Rint | 0.124 | 0.058 | 0.095 | 0.039 | 0.027 |
| R[F2 > 2σ(F2)] a | 0.0743 | 0.0799 | 0.0517 | 0.0389 | 0.0441 |
| wR2 [all data] b | 0.2205 | 0.1650 | 0.1220 | 0.0861 | 0.1248 |
| Largest difference map features/eÅ−3 | 1.40, −0.49 | 0.46, −0.52 | 0.38, −0.30 | 0.29, −0.16 | 0.51, −0.21 |
| 1a | 1b·CH3OH | 2f·CH3OH a | 2g | 3 | |
|---|---|---|---|---|---|
| O–H···Nintra | 2.745(4), 119(4) | ||||
| O–HMeOH···Ointer O–HMeOH···OMeOH | 2.844(8), 157 2.781(11), 172 | ||||
| O–H···Pinter | 3.432(3), 173 | 3.4400(12), 167(2) | |||
| O–H···(O)Cinter | 2.671(3), 171(3) [2.659(3), 165(3)] | 2.706(4), 169(4) | |||
| N–H···Nintra | 2.695(3), 114(2) [2.714(3), 117(2)] | 2.748(4), 114(3) |
| Com-pound a | δ(P) b | δ(H) /OH (NH) | δ(H) /arom. H. | δ(H) /CH2 | δ(H) /CH2 | δ(H) /CH3 | νOH (νNH) f | νPtCl | Microanalysis (CHN) |
|---|---|---|---|---|---|---|---|---|---|
| 4a (98) | −9.4 d (3424) | 9.25 | 7.89–7.80, 7.64–7.46, 6.68, 5.90 | 4.21 | 1.93 | 3314 | 316, 289 | Calc. for C33H31Cl2NOP2Pt.CH2Cl2, C, 46.91; H, 3.82; N, 1.61 Found, C, 47.07; H, 3.77; N, 1.69 | |
| 4b (89) | −4.9 d (3426) | 9.22 | 7.96–7.53, 6.96, 6.49, 6.33 | 4.19 | 1.29 | 3373 | 315, 282 | Calc. for C33H31Cl2NOP2Pt, C, 50.46; H, 3.98; N, 1.78 Found, C, 50.51; H, 4.13; N, 1.83 | |
| 4c (78) | −8.6 d (3436) | 9.42 | 7.89–7.84, 7.56–7.43, 6.59, 6.30, 6.05 | 4.16 | 2.06 | 3433 | 309, 290 | Calc. for C33H31Cl2NOP2Pt.0.5CH2Cl2, C, 48.96; H, 3.87; N, 1.68 Found, C, 49.42; H, 3.96; N, 1.73 | |
| 4d (98) | −11.7 d (3410) | 8.44 | 7.94–7.87, 7.78–7.62, 6.86, 6.47 | 4.43 | 2.02 | 3421 | 314, 290 | Calc. for C33H31Cl2NOP2Pt, C, 50.46; H, 3.98; N, 1.78 Found, C, 50.24; H, 3.98; N, 1.85 | |
| 4e (81) | −7.8 d (3421) | 9.01 | 7.96–7.85, 7.59–7.45, 6.75, 6.27, 6.03 | 4.33 | 2.09 | 3416 | 316, 284 | Calc. for C33H31Cl2NOP2Pt, C, 50.46; H, 3.98; N, 1.78 Found, C, 50.66; H, 4.61; N, 1.70 | |
| 5a (89) | −9.8 d,e (3398) | 9.45 (8.91) | 7.84–7.80, 7.53–7.44, 6.69 | 3.49 | 4.05 | 2.22 | 3051 (3249) | 305, 283 | Calc. for C35H34Cl2N2O2P2Pt.0.5CH2Cl2, C, 48.63; H, 3.74; N, 3.15 Found, C, 49.00; H, 4.07; N, 3.13 |
| 5b (65) | −11.0 d (3397) | 9.16 (8.61) | 7.83–7.80, 7.57–7.41, 7.05, 6.48 | 4.03 | 4.03 | 1.80 | 3050 (3350) | 316, 283 | Calc. for C35H34Cl2N2O2P2Pt, C, 49.89; H, 4.07; N, 3.32 Found, C, 49.32; H, 4.17; N, 3.25 |
| 5c (73) | −9.9 d (3405) | 9.56 (8.94) | 7.85–7.77, 7.59–7.38, 6.63, 6.51 | 3.17 | 4.05 | 2.17 | 3075 (3347) | 315, 290 | Calc. for C35H34Cl2N2O2P2Pt, C, 49.89; H, 4.07; N, 3.32 Found, C, 49.28; H, 4.05; N, 2.91 |
| 5d (99) | −9.8 c,d (3406) | 9.17 (8.90) | 7.98–7.50, 6.97–6.84, 6.68, 6.73 | 3.20 | 4.66 | 2.13 | 3323 (3465) | 309, 283 | Calc. for C35H34Cl2N2O2P2Pt.0.5C4H10O, C, 50.52; H, 4.47; N, 3.19 Found, C, 50.91; H, 4.53; N, 3.61 |
| 5e (90) | −9.7 c,d (3406) | 9.46 (9.21) | 7.94–7.78, 7.54–7.42, 7.09, 6.87, 6.69 | 3.43 | 4.12 | 2.02 | 3287 (3439) | 312, 286 | Calc. for C35H34Cl2N2O2P2Pt, C, 49.89; H, 4.07; N, 3.32 Found, C, 49.77; H, 3.95; N, 3.38 |
| 5f (85) | −9.5 c,d (3425) | 9.62 (9.36) | 7.91–7.86, 7.60–7.42, 7.05, 6.83, 6.45 | 3.47 | 4.18 | 3053 (3312) | 304, 279 | Calc. for C34H32Cl2N2O2P2Pt, C, 49.29; H, 3.89; N, 3.38 Found, C, 48.98; H, 3.38; N, 3.37 | |
| 5g (84) | −9.5 c,d (3405) | 9.52 (9.31) | 8.01–7.97, 7.70–7.61, 7.34, 6.78 | 3.49 | 4.26 | 3054 (3325) | 311, 287 | Calc. for C34H32Cl2N2O2P2Pt, C, 49.29; H, 3.89; N, 3.38 Found, C, 48.72; H, 3.66; N, 3.33 | |
| 6 (89) | −4.0 d (3436) | 8.45 | 7.45–7.05, 6.89–6.76, 6.31, | 4.31 | 3356 | 311, 289 | Calc. for C32H29Cl2NOP2Pt, C, 49.82; H, 3.79; N, 1.82 Found, C, 49.31; H, 3.58; N, 1.79 |
| Compound | 4b·(CH3)2SO | 4c·CHCl3 | 4d·½OEt2 | 4e·½CHCl3·½CH3OH |
|---|---|---|---|---|
| Formula | C35H37Cl2NO2P2PtS | C34H32Cl5NOP2Pt | C35H36Cl2NO1.5P2Pt | C34H33.5Cl3.5NO1.5P2Pt |
| M | 863.64 | 904.88 | 822.58 | 861.22 |
| Crystal dimensions | 0.19 × 0.02 × 0.01 | 0.30 × 0.18 × 0.04 | 0.13 × 0.06 × 0.03 | 0.15 × 0.04 × 0.02 |
| Crystal morphology and colour | Needle, colourless | Plate, colourless | Lath, colourless | Needle, colourless |
| Crystal system | Tetragonal | Monoclinic | Monoclinic | Monoclinic |
| Space group | P43 | P21/n | P21/c | P21/n |
| a/Å | 11.373(3) | 11.6938(4) | 15.7344(6) | 21.4521(4) |
| b/Å | 16.7052(6) | 17.0714(6) | 12.5164(2) | |
| c/Å | 26.773(6) | 18.2242(7) | 13.9632(5) | 24.5837(4) |
| α/° | ||||
| β/° | 99.7066(6) | 92.0800(4) | 92.2343(5) | |
| γ/° | ||||
| V/Å3 | 3463(2) | 3509.1(2) | 3748.2(2) | 6595.78(19) |
| Z | 4 | 4 | 4 | 8 |
| λ/Å | 0.71073 | 0.71073 | 0.6710 | 0.71073 |
| Τ/Κ | 150(2) | 150(2) | 150(2) | 120(2) |
| Density (calcd.)/Mg/m3 | 1.657 | 1.713 | 1.458 | 1.735 |
| μ/mm−1 | 4.391 | 4.500 | 3.425 | 4.666 |
| θ range/° | 1.79–26.09 | 1.67–31.09 | 1.78–31.10 | 2.94–27.49 |
| Measured reflections | 29,848 | 32,600 | 48,268 | 84,353 |
| Independent reflections | 6852 | 10,997 | 13,239 | 15,063 |
| Observed reflections (F2 > 2σ(F2)) | 5560 | 8926 | 10,918 | 12,905 |
| Rint | 0.110 | 0.043 | 0.039 | 0.049 |
| R[F2 > 2σ(F2)] a | 0.0473 | 0.0303 | 0.0266 | 0.0561 |
| wR2 [all data] b | 0.1015 | 0.0660 | 0.0646 | 0.1202 |
| Largest difference map features/eÅ−3 | 1.43, −0.91 | 1.29, −1.08 | 0.84, −0.67 | 1.64, −1.48 |
| Compound | 5a·½OEt2 | 5b | 5c·¼H2O | 5d·OEt2 | 6·(CH3)2SO |
|---|---|---|---|---|---|
| Formula | C37H39Cl2N2O2.50P2Pt | C35H34Cl2N2O2P2Pt | C35H34.5Cl2N2O2.25P2Pt | C39H44Cl2N2O3P2Pt | C34H35Cl2NO2P2PtS |
| M | 879.63 | 842.57 | 842.57 | 916.69 | 849.62 |
| Crystal dimensions | 0.12 × 0.06 × 0.05 | 0.05 × 0.02 × 0.01 | 0.09 × 0.05 × 0.02 | 0.13 × 0.12 × 0.02 | 0.32 × 0.11 × 0.02 |
| Crystal morphology and colour | Block, colourless | Plate, colourless | Plate, colourless | Plate, colourless | Needle, colourless |
| Crystal system | Trigonal | Monoclinic | Triclinic | Orthorhombic | Monoclinic |
| Space group | P32 | P21/n | P | Pbcm | P21/n |
| a/Å | 24.3688(7) | 18.2384(7) | 8.4021(6) | 10.125(6) | 9.7763(4) |
| b/Å | 8.1955(3) | 10.3896(7) | 19.790(11) | 13.0930(5) | |
| c/Å | 10.6567(6) | 23.5809(10) | 21.8810(15) | 18.407(10) | 25.8715(10) |
| α/° | 92.8380(10) | ||||
| β/° | 111.4543(5) | 97.9841(9) | 95.1690(6) | ||
| γ/° | 106.6253(8) | ||||
| V/Å3 | 5480.5(4) | 3280.5(2) | 1804.5(2) | 3688(4) | 3298.1(2) |
| Z | 6 | 4 | 2 | 4 | 4 |
| λ/Å | 0.7848 | 0.6910 | 0.6710 | 0.71073 | 0.71073 |
| Τ/Κ | 150(2) | 120(2) | 150(2) | 150(2) | 150(2) |
| Density (calcd.)/Mg/m3 | 1.599 | 1.706 | 1.559 | 1.651 | 1.711 |
| μ/mm−1 | 5.266 | 4.225 | 3.561 | 4.077 | 4.609 |
| θ range/° | 3.69–33.17 | 1.19–31.01 | 2.71–30.94 | 2.01–25.00 | 1.58–30.64 |
| Measured reflections | 48,822 | 37,553 | 22,910 | 25,092 | 38,887 |
| Independent reflections | 19,298 | 10,642 | 12,184 | 3357 | 10,104 |
| Observed reflections (F2 > 2σ(F2)) | 17,145 | 8283 | 10,104 | 2051 | 7753 |
| Rint | 0.071 | 0.063 | 0.053 | 0.1504 | 0.0572 |
| R[F2 > 2σ(F2)] a | 0.0542 | 0.0363 | 0.0507 | 0.0746 | 0.0356 |
| wR2 [all data] b | 0.1551 | 0.0842 | 0.1341 | 0.2065 | 0.0816 |
| Largest difference map features/eÅ−3 | 1.59, −1.71 | 1.40, −1.47 | 2.34, −3.46 | 2.77, −1.91 | 1.97, −1.50 |
| Bond Length (Å) | 4b·(CH3)2SO | 4c·CHCl3 | 4d | 4e·½CHCl3·½MeOH a |
|---|---|---|---|---|
| Pt(1)–P(1) | 2.223(4) | 2.2226(6) | 2.2257(6) | 2.2386(18) [2.2353(18)] |
| Pt(1)–P(2) | 2.225(4) | 2.2186(7) | 2.2146(6) | 2.2475(18) [2.2464(18)] |
| Pt(1)–Cl(1) | 2.358(4) | 2.3625(6) | 2.3558(6) | 2.3560(18) [2.3574(17)] |
| Pt(1)–Cl(2) | 2.359(4) | 2.3484(6) | 2.3553(6) | 2.3694(17) [2.3616(18)] |
| Bond angle (°) | ||||
| Cl(1)–Pt(1)–P(1) | 87.91(14) | 86.12(2) | 87.63(2) | 87.30(7) [86.87(7)] |
| Cl(1)–Pt(1)–P(2) | 174.97(15) | 176.08(3) | 175.93(2) | 176.50(7) [176.84(7)] |
| Cl(1)–Pt(1)–Cl(2) | 88.95(13) | 88.68(2) | 90.43(2) | 88.13(7) [87.34(7)] |
| Cl(2)–Pt(1)–P(2) | 86.71(13) | 88.94(2) | 85.53(2) | 88.54(7) [90.04(7)] |
| Cl(2)–Pt(1)–P(1) | 176.37(15) | 174.73(2) | 177.81(2) | 169.14(7) [170.73(7)] |
| P(1)–Pt(1)–P(2) | 96.35(14) | 96.30(3) | 96.42(2) | 96.17(7) [95.95(7)] |
| Bond Length (Å) | 5a·½OEt2 a | 5b | 5c·¼H2O | 5d·OEt2 | 6·(CH3)2SO |
|---|---|---|---|---|---|
| Pt(1)–P(1) | 2.233(3) [2.234(3)] | 2.2172(9) | 2.2268(12) | 2.220(3) | 2.2219(9) |
| Pt(1)–P(2) | 2.230(3) [2.229(3)] | 2.2249(9) | 2.2196(12) | c | 2.2288(9) |
| Pt(1)–Cl(1) | 2.381(3) [2.378(3)] | 2.3685(9) | 2.347(4) b | 2.348(3) | 2.3421(9) |
| Pt(1)–Cl(2) | 2.361(3) [2.365(3) | 2.3425(9) | 2.3638(12) | c | 2.3618(10) |
| Bond angle (°) | |||||
| Cl(1)–Pt(1)–P(1) | 86.31(10) [86.48(10)] | 85.73(3) | 92.55(11) | 89.98(12) | 88.81(3) |
| Cl(1)–Pt(1)–P(2) | 177.81(12) [177.51(12)] | 176.29(3) | 167.1(2) | 176.96(13) | 173.98(3) |
| Cl(1)–Pt(1)–Cl(2) | 90.20(13) [90.16(13)] | 89.33(3) | 88.17(11) | 87.38(17) | 88.78(4) |
| Cl(2)–Pt(1)–P(2) | 87.75(14) [87.51(13)] | 88.31(3) | 87.76(4) | 89.98(12) | 87.17(3) |
| Cl(2)–Pt(1)–P(1) | 175.76(12) [175.67(12)] | 174.72(3) | 178.70(5) | 176.96(13) | 174.52(4) |
| P(1)–Pt(1)–P(2) | 90.20(13) [95.81(11)] | 96.52(3) | 91.30(4) | 92.62(17) | 94.83(3) |
| 4b·(CH3)2SO | 4c·CHCl3 | 4e·½CHCl3 ·½CH3OH | 5a·½OEt2 a | 5b | 5c·¼H2O | 5d·OEt2 | 6·(CH3)2SO | |
|---|---|---|---|---|---|---|---|---|
| O–H···(O)Cinter | 3.714(14), 169 | |||||||
| N–H···Nintra | 2.711(5), 107(4) | 2.776(12), 108 b | ||||||
| O–Hinter···OMeOH | 2.624(10), 160 | |||||||
| O–Hinter···ClPt | 3.145(2), 145(3) 3.361(2), 133(3) | 3.197(6), 160(9) | 3.065(3), 161(5) | |||||
| O–Hinter···O(CH3)2SO | 2.716(17), 170 | 1.79(2), 173(5) | ||||||
| N–Hinter···ClPt | 3.328(12), 145(16) 3.320(11), 159(16) | 3.505(15), 138(6) | ||||||
| O–H···(O)Cintra | 2.596(13), 157 2.610(13), 175(20) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De’Ath, P.; Elsegood, M.R.J.; Sanchez-Ballester, N.M.; Smith, M.B. Low-Dimensional Architectures in Isomeric cis-PtCl2{Ph2PCH2N(Ar)CH2PPh2} Complexes Using Regioselective-N(Aryl)-Group Manipulation. Molecules 2021, 26, 6809. https://doi.org/10.3390/molecules26226809
De’Ath P, Elsegood MRJ, Sanchez-Ballester NM, Smith MB. Low-Dimensional Architectures in Isomeric cis-PtCl2{Ph2PCH2N(Ar)CH2PPh2} Complexes Using Regioselective-N(Aryl)-Group Manipulation. Molecules. 2021; 26(22):6809. https://doi.org/10.3390/molecules26226809
Chicago/Turabian StyleDe’Ath, Peter, Mark R. J. Elsegood, Noelia M. Sanchez-Ballester, and Martin B. Smith. 2021. "Low-Dimensional Architectures in Isomeric cis-PtCl2{Ph2PCH2N(Ar)CH2PPh2} Complexes Using Regioselective-N(Aryl)-Group Manipulation" Molecules 26, no. 22: 6809. https://doi.org/10.3390/molecules26226809
APA StyleDe’Ath, P., Elsegood, M. R. J., Sanchez-Ballester, N. M., & Smith, M. B. (2021). Low-Dimensional Architectures in Isomeric cis-PtCl2{Ph2PCH2N(Ar)CH2PPh2} Complexes Using Regioselective-N(Aryl)-Group Manipulation. Molecules, 26(22), 6809. https://doi.org/10.3390/molecules26226809