
Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives



Abstract
:
1. Introduction
2. Staphylococcus aureus Efflux Pump Inhibitors
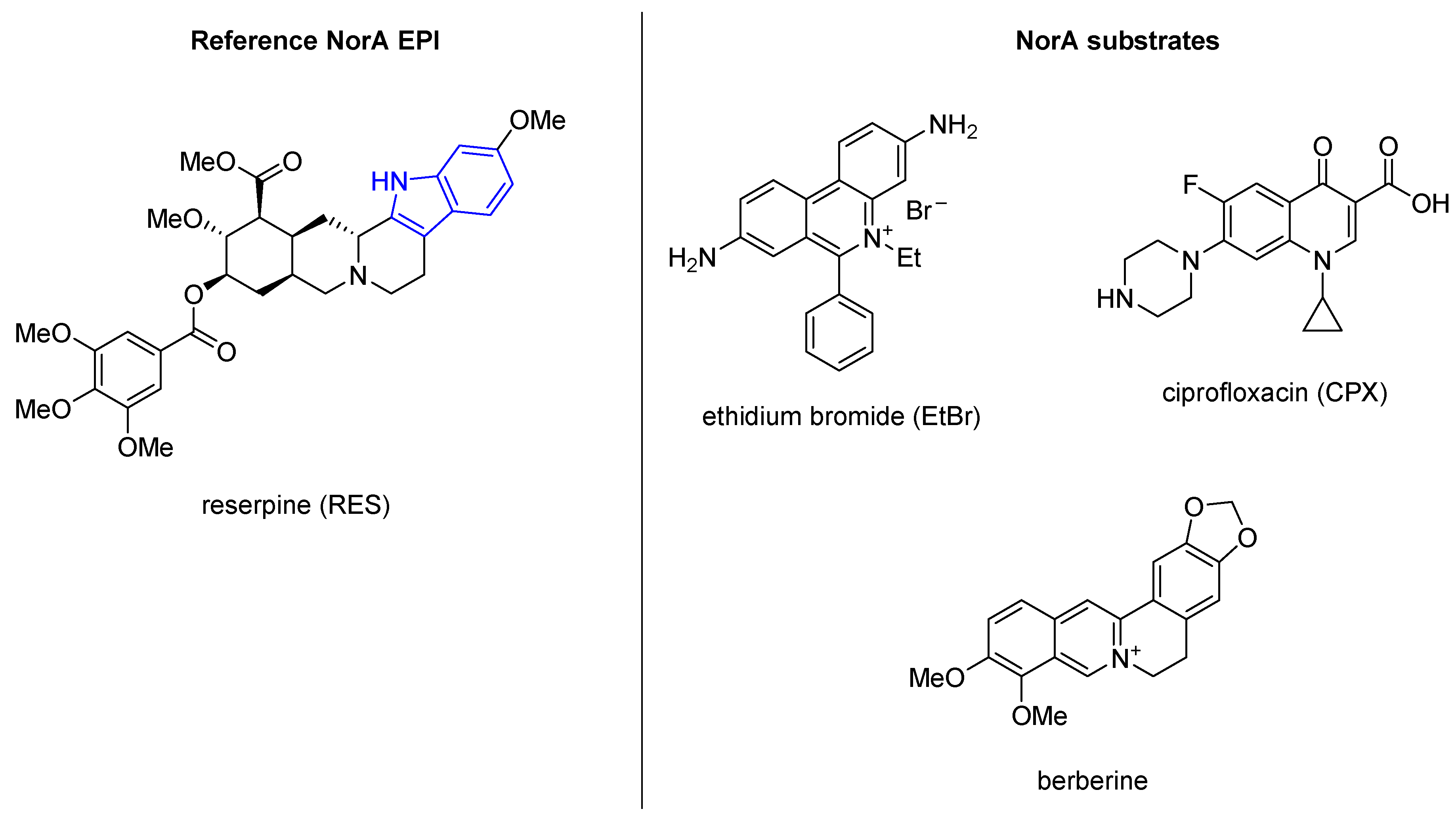
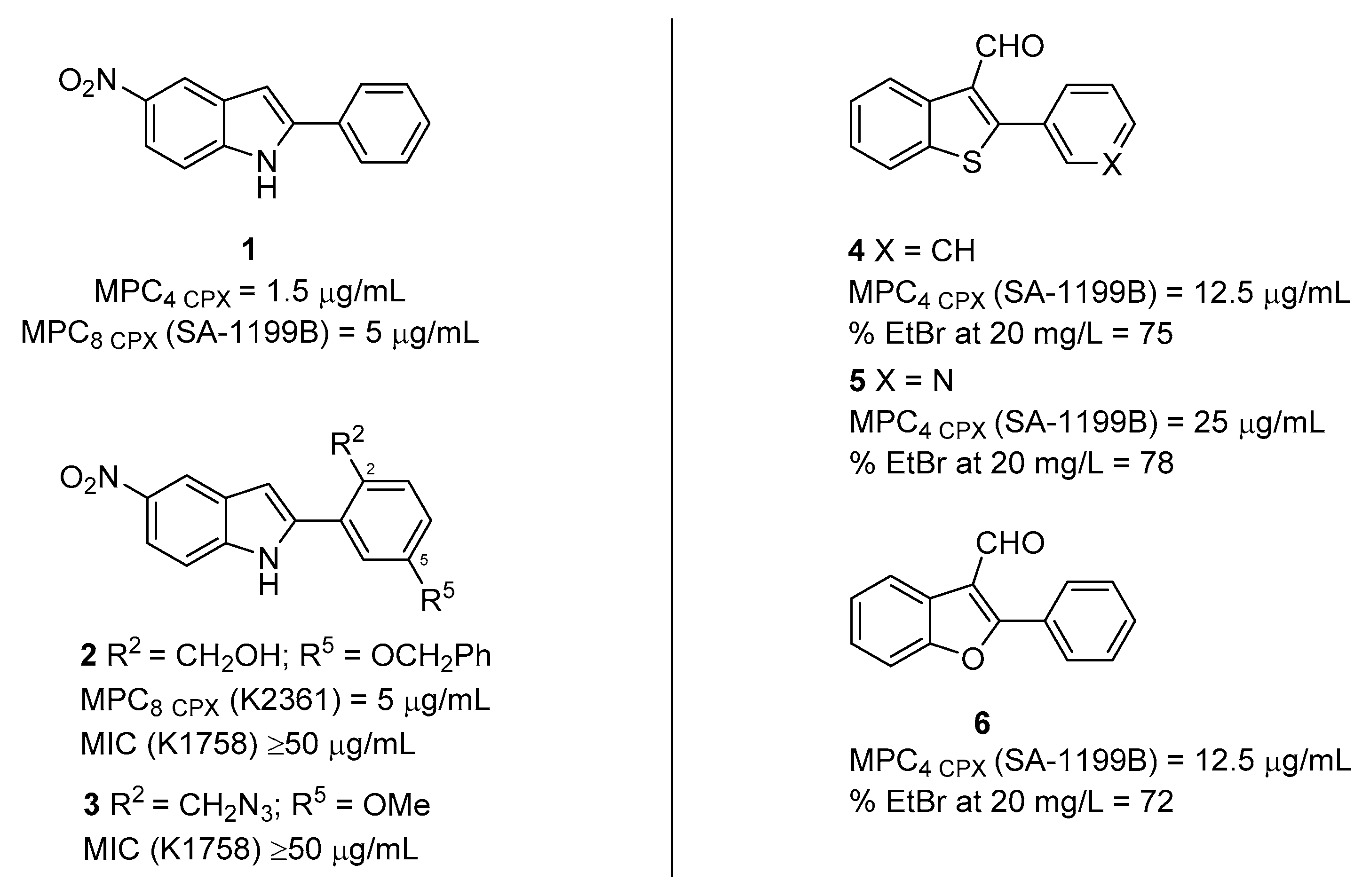
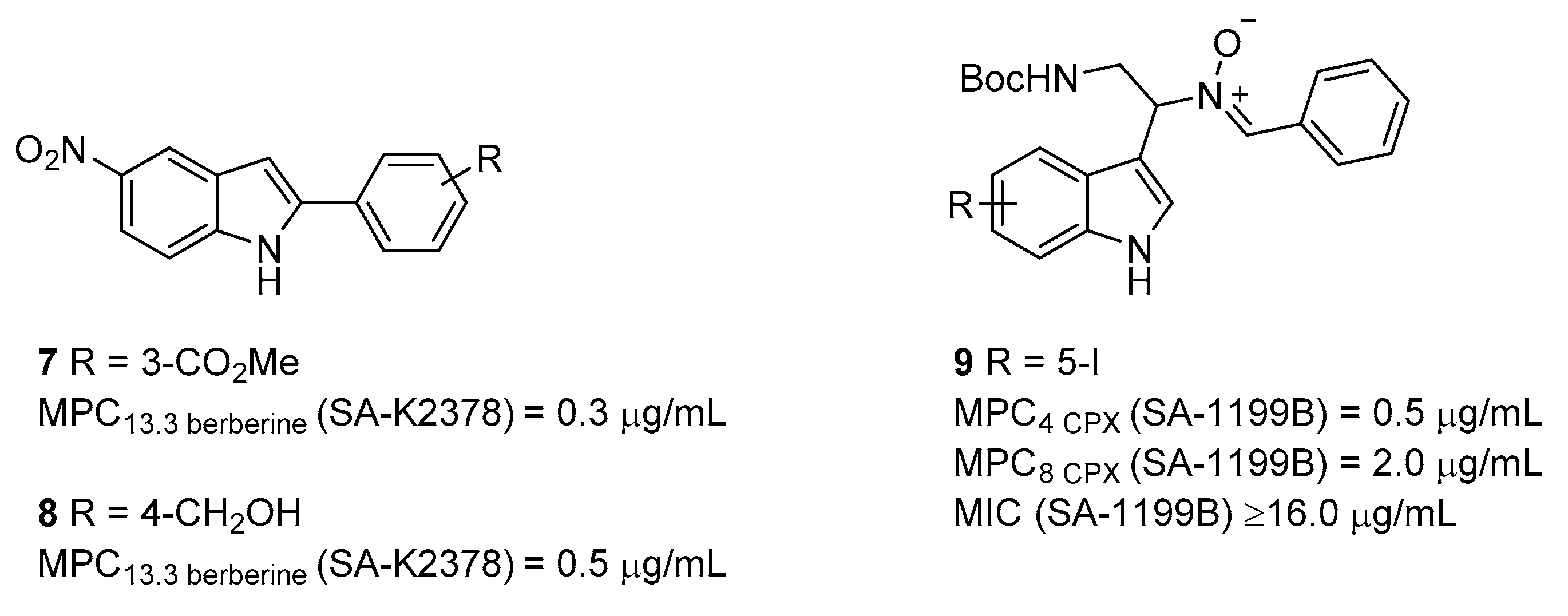
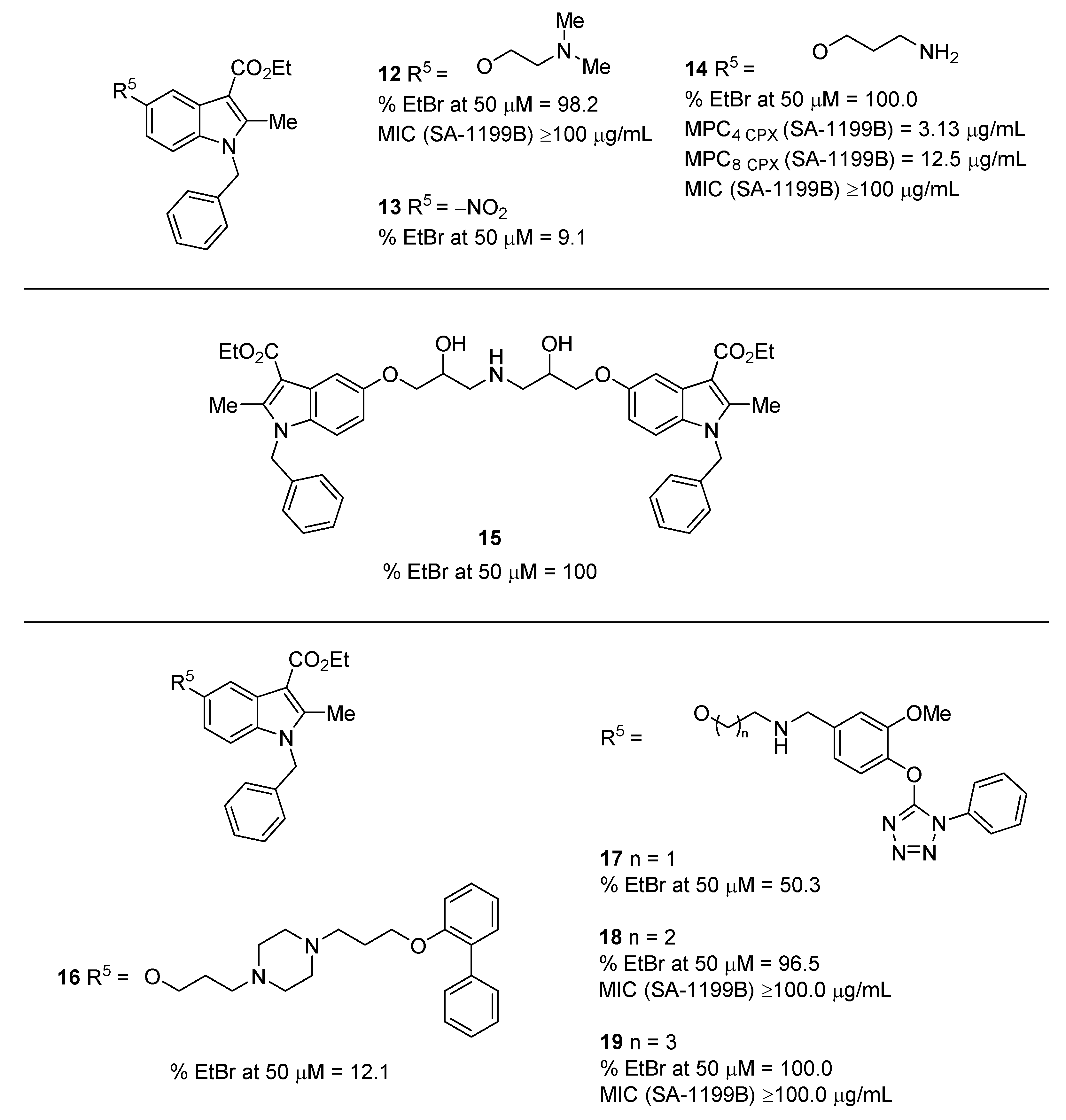
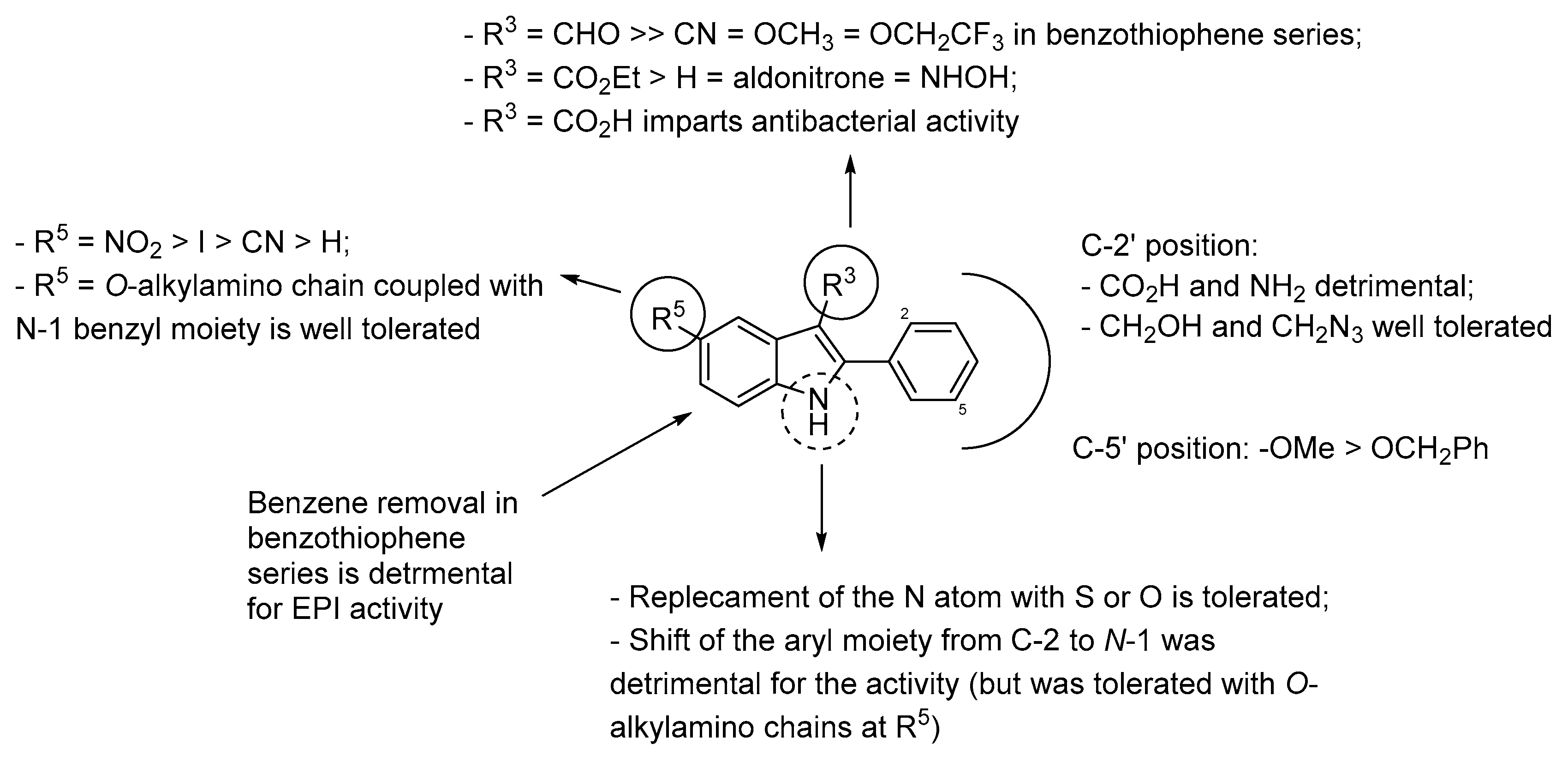
2.1. Indole Derivatives
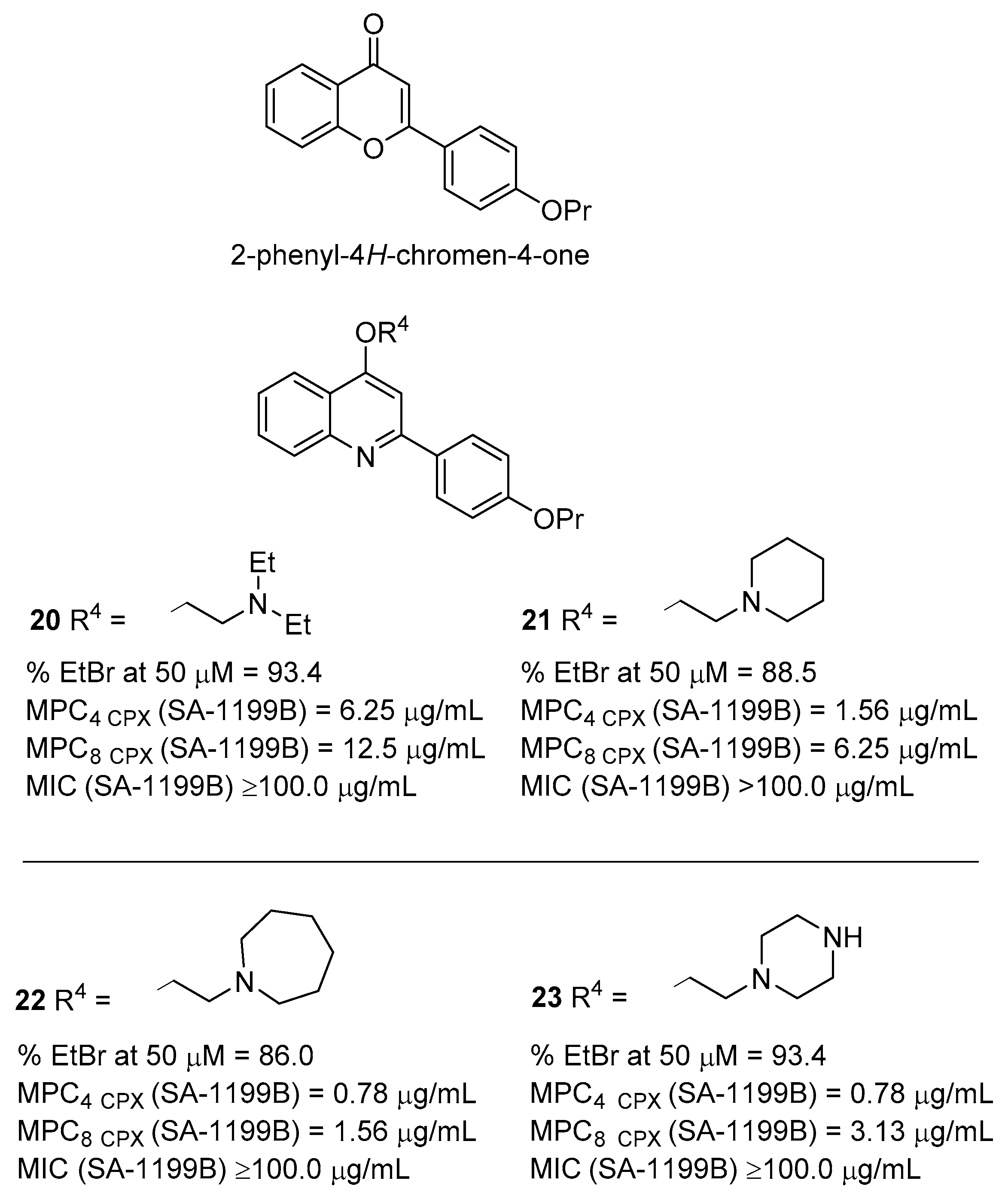
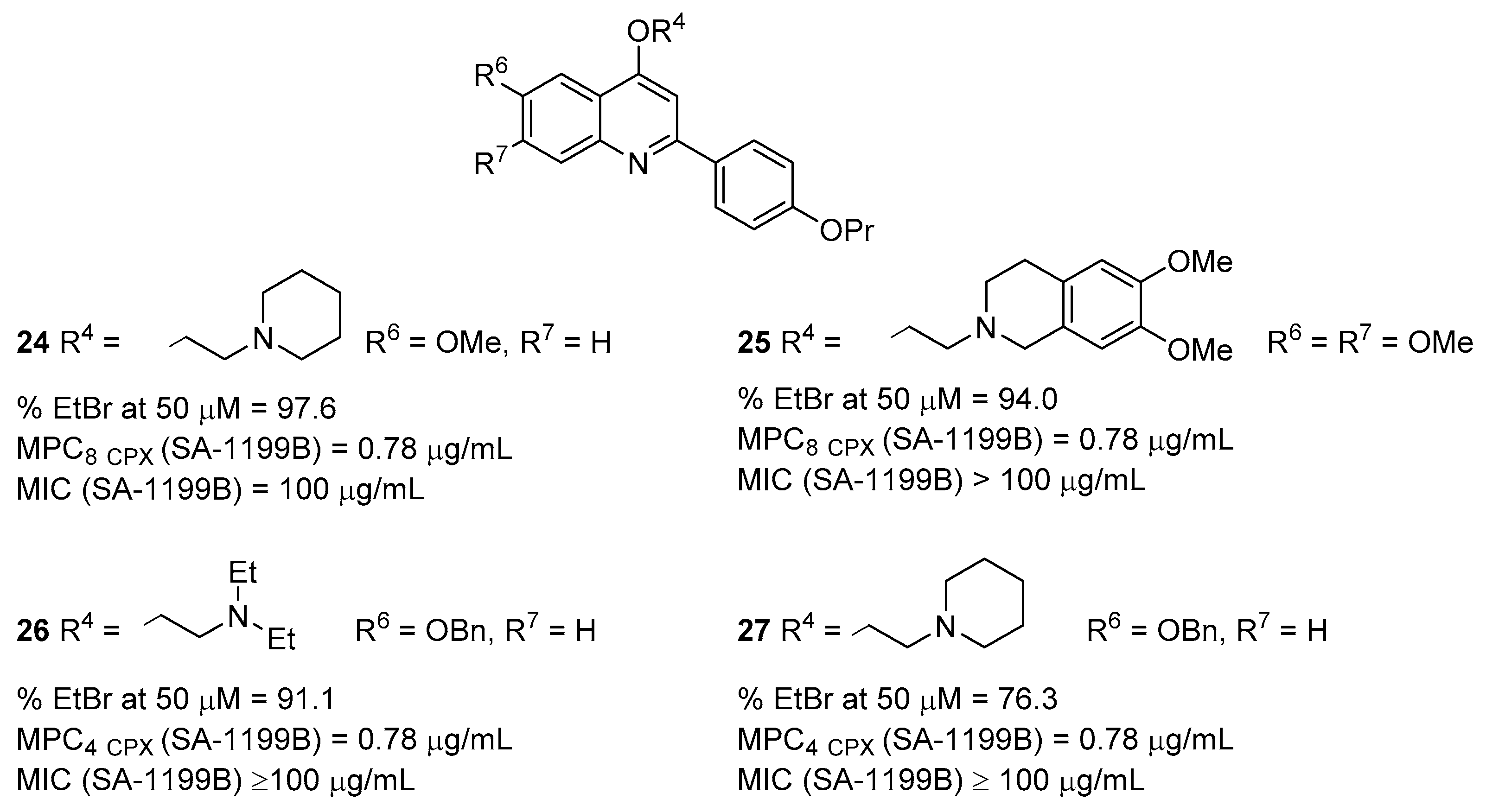
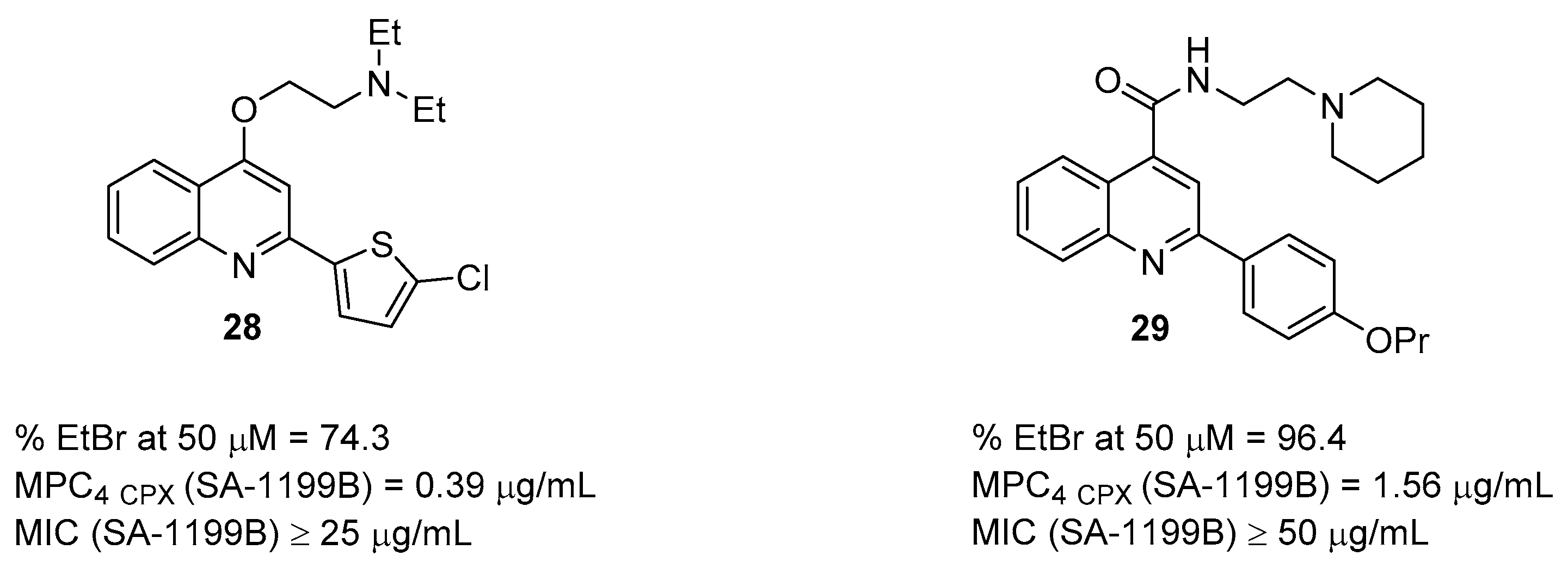
2.2. Quinoline Derivatives
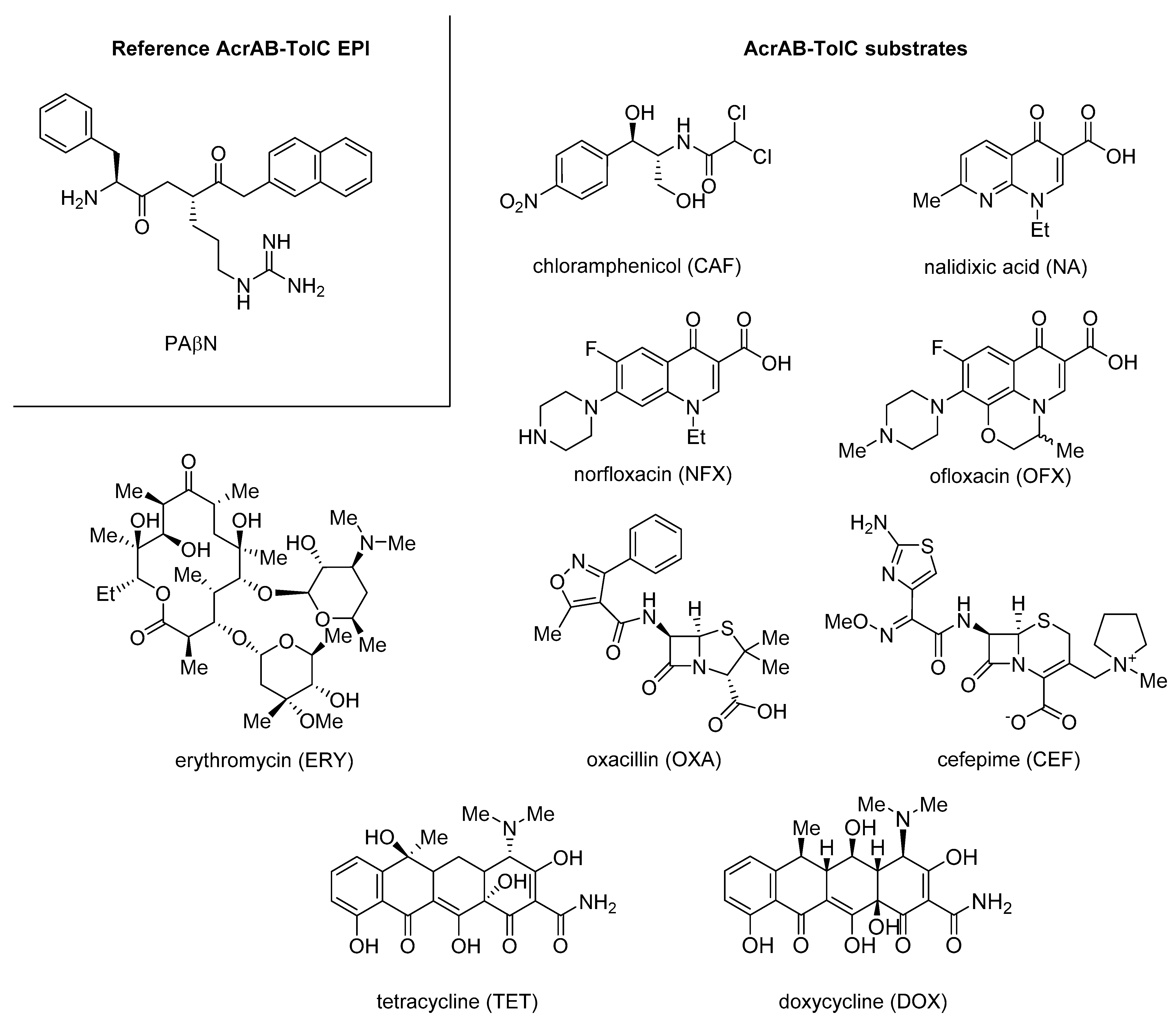
3. Gram-Negative Efflux Pump Inhibitors
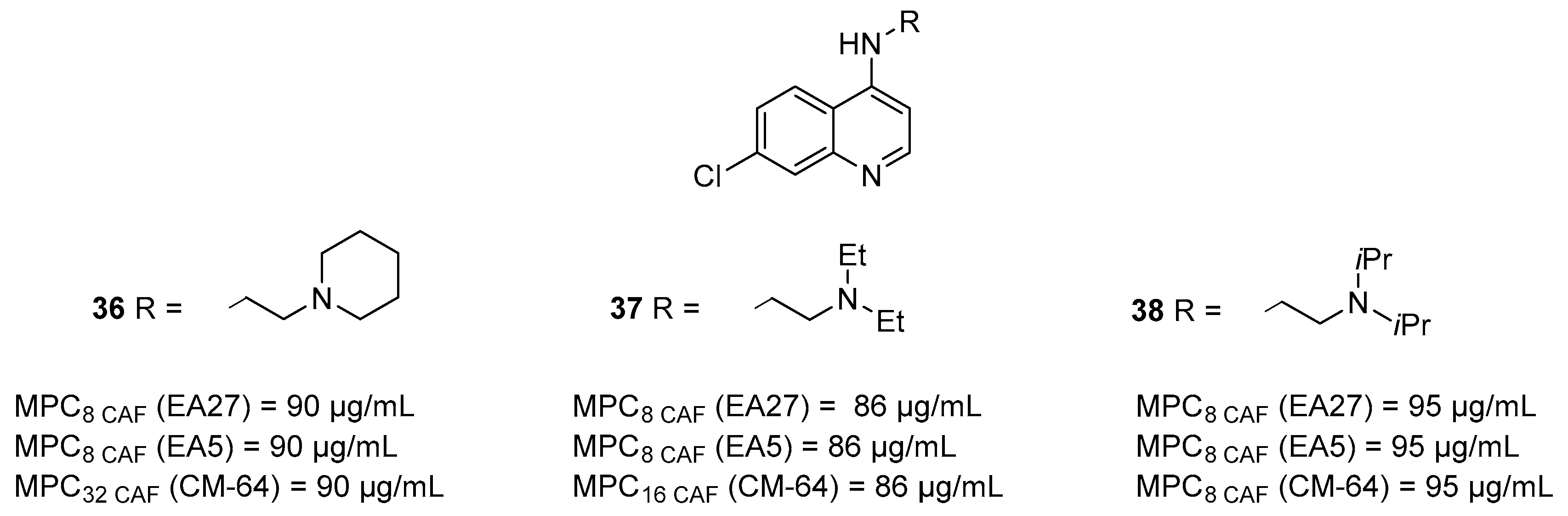
3.1. Quinoline Derivatives
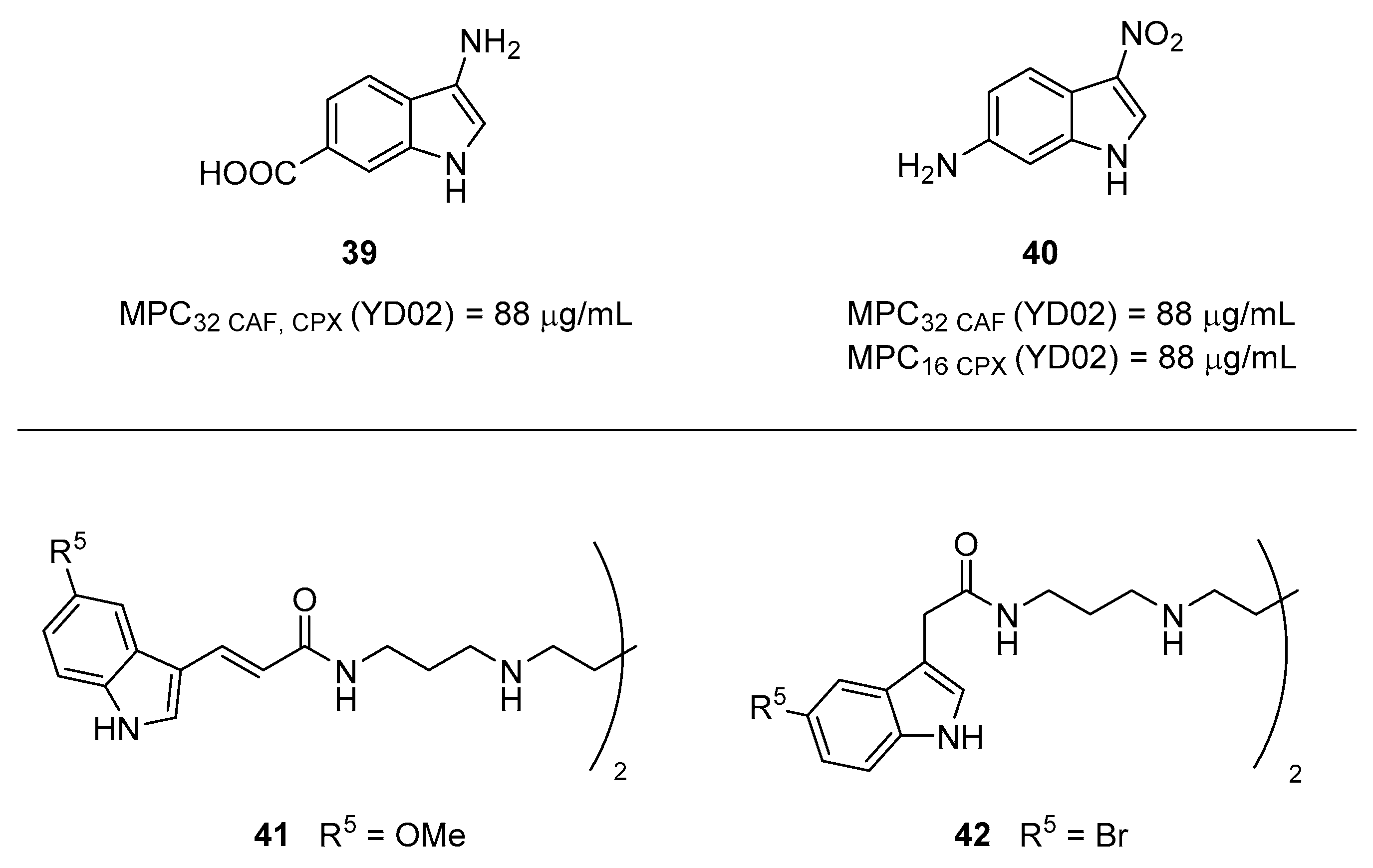
3.2. Indole Derivatives
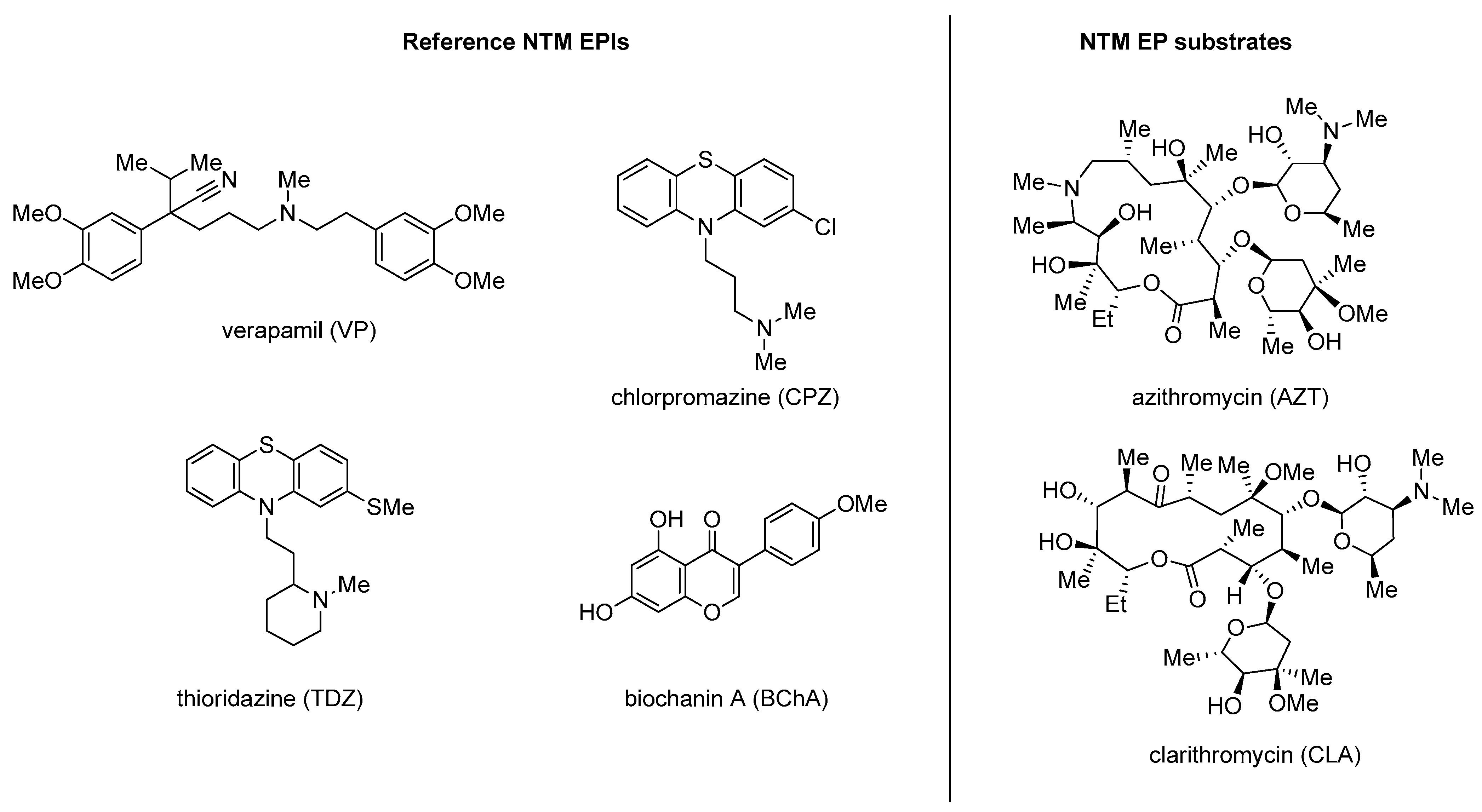
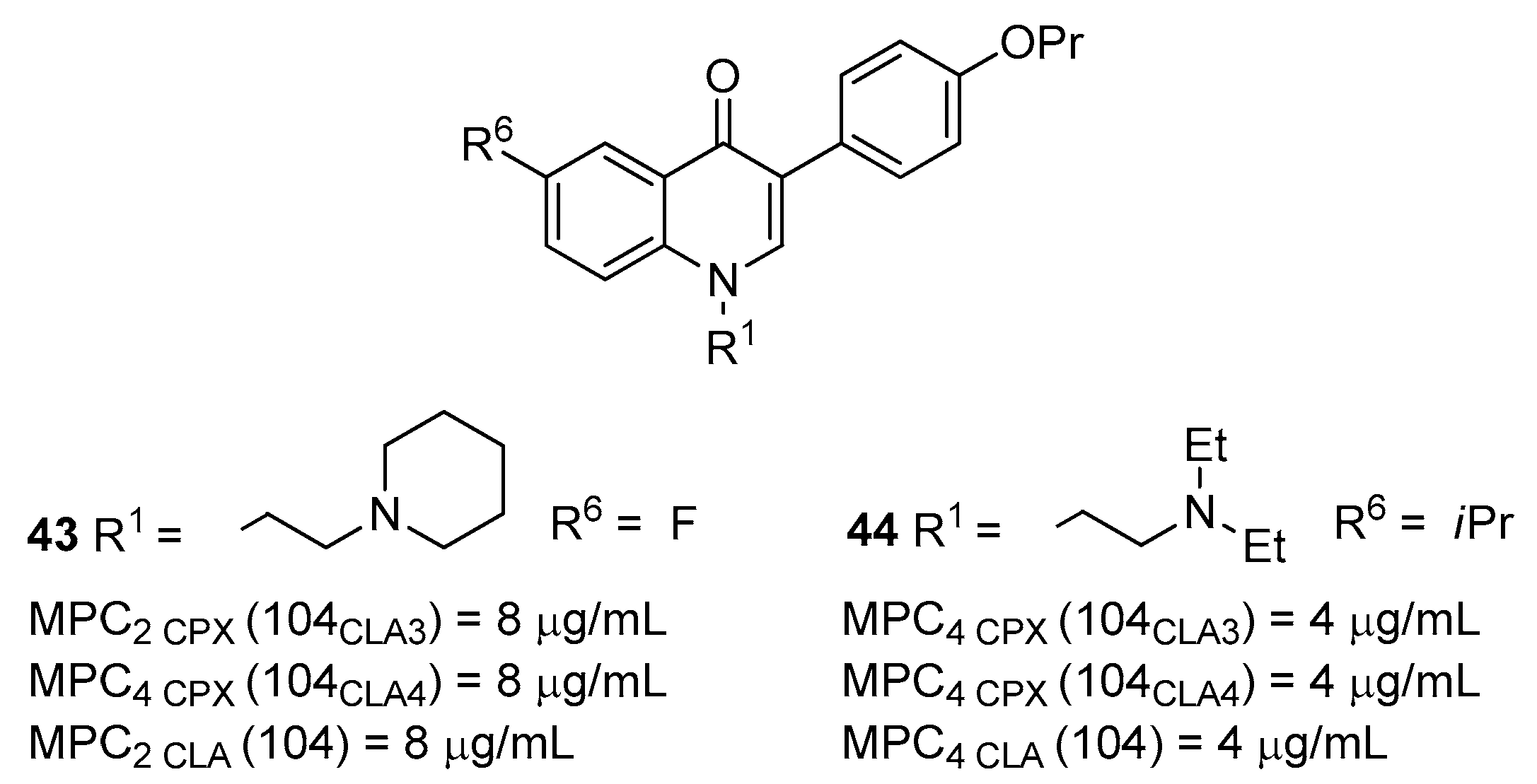
4. Nontuberculous Mycobacteria (NTM) Efflux Pump Inhibitors
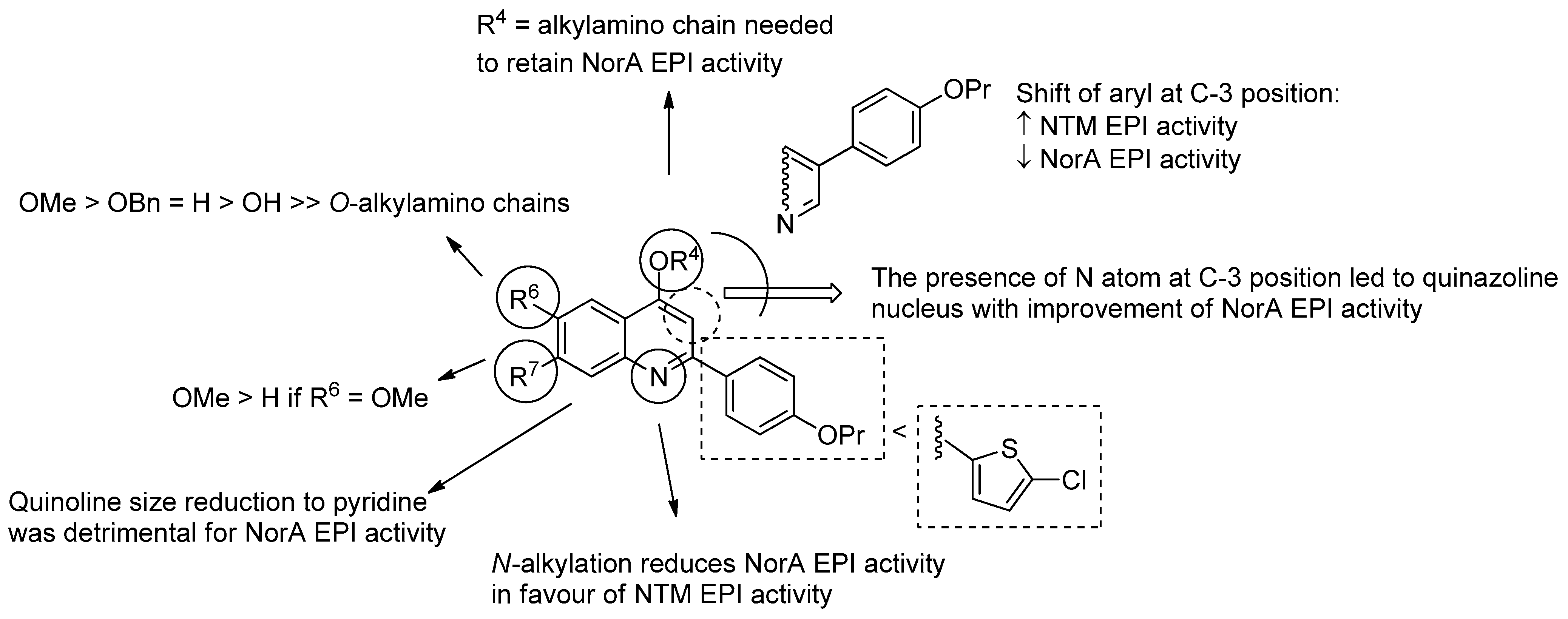
Quinoline Derivatives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- World Health Organization. Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report: 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Miethke, M.; Pieroni, M.; Weber, T.; Brönstrup, M.; Hammann, P.; Halby, L.; Arimondo, P.B.; Glaser, P.; Aigle, B.; Bode, H.B.; et al. Towards the sustainable discovery and development of new antibiotics. Nat. Rev. Chem. 2021, 5, 726–749. [Google Scholar] [CrossRef] [PubMed]
- Kyriakides, S. EU Action on Antimicrobial Resistance|Public Health. 2016. Available online: https://ec.europa.eu/health/antimicrobial-resistance/eu-action-on-antimicrobial-resistance.it (accessed on 16 November 2021).
- Melander, R.J.; Melander, C. The Challenge of Overcoming Antibiotic Resistance: An Adjuvant Approach? ACS Infect. Dis. 2017, 3, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Brown, D. Antibiotic resistance breakers: Can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 2015, 14, 821–832. [Google Scholar] [CrossRef]
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; Dastidar, S.G.; Fanning, S.; Kristiansen, J.E.; Molnar, J.; Pagès, J.-M.; Schelz, Z.; Spengler, G.; Viveiros, M.; Amaral, L. Potential role of non-antibiotics (helper compounds) in the treatment of multidrug-resistant Gram-negative infections: Mechanisms for their direct and indirect activities. Int. J. Antimicrob. Agents 2008, 31, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Rampioni, G.; Pillai, C.R.; Longo, F.; Bondì, R.; Baldelli, V.; Messina, M.; Imperi, F.; Visca, P.; Leoni, L. Effect of efflux pump inhibition on Pseudomonas aeruginosa transcriptome and virulence. Sci. Rep. 2017, 7, 11392. [Google Scholar] [CrossRef]
- Vermote, A.; Van Calenbergh, S. Small-Molecule Potentiators for Conventional Antibiotics against Staphylococcus aureus. ACS Infect. Dis. 2017, 3, 780–796. [Google Scholar] [CrossRef]
- González-Bello, C.; Rodríguez, D.; Pernas, M.; Rodríguez, Á.; Colchón, E. β-Lactamase Inhibitors to Restore the Efficacy of Antibiotics against Superbugs. J. Med. Chem. 2020, 63, 1859–1881. [Google Scholar] [CrossRef] [PubMed]
- Piddock, L.J.V. Multidrug-resistance efflux pumps—Not just for resistance. Nat. Rev. Microbiol. 2006, 4, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef]
- Saier, M.H.; Tran, C.V.; Barabote, R.D. TCDB: The Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006, 34, 181–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, B.D.; Kaatz, G.W. Multidrug efflux pumps of Gram-positive bacteria. Drug Resist. Updates 2016, 27, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Venter, H.; Mowla, R.; Ohene-Agyei, T.; Ma, S. RND-type drug efflux pumps from Gram-negative bacteria: Molecular mechanism and inhibition. Front. Microbiol. 2015, 6, 377. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Bostian, K.A. Practical applications and feasibility of efflux pump inhibitors in the clinic—A vision for applied use. Biochem. Pharmacol. 2006, 71, 910–918. [Google Scholar] [CrossRef]
- Piddock, L.J.V. Understanding the basis of antibiotic resistance: A platform for drug discovery. Microbiology 2014, 160, 2366–2373. [Google Scholar] [CrossRef] [Green Version]
- Ricci, V.; Tzakas, P.; Buckley, A.; Coldham, N.C.; Piddock, L.J.V. Ciprofloxacin-resistant Salmonella enterica serovar typhimurium strains are difficult to select in the absence of AcrB and TolC. Antimicrob. Agents Chemother. 2006, 50, 38–42. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lambert, G.; Liao, D.; Kim, H.; Robin, K.; Tung, C.K.; Pourmand, N.; Austin, R.H. Acceleration of emergence of bacterial antibiotic resistance in connected microenvironments. Science 2011, 333, 1764–1767. [Google Scholar] [CrossRef] [PubMed]
- Baugh, S.; Phillips, C.R.; Ekanayaka, A.S.; Piddock, L.J.V.; Webber, M.A. Inhibition of multidrug efflux as a strategy to prevent biofilm formation. J. Antimicrob. Chemother. 2014, 69, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Kvist, M.; Hancock, V.; Klemm, P. Inactivation of efflux pumps abolishes bacterial biofilm formation. Appl. Environ. Microbiol. 2008, 74, 7376–7382. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, S.; Piccioni, M.; Felicetti, T.; De Marco, S.; Manfroni, G.; Pagiotti, R.; Nocchetti, M.; Cecchetti, V.; Pietrella, D. Investigation on the effect of known potent: S. aureus NorA efflux pump inhibitors on the staphylococcal biofilm formation. RSC Adv. 2017, 7, 37007–37014. [Google Scholar] [CrossRef] [Green Version]
- Schillaci, D.; Spanò, V.; Parrino, B.; Carbone, A.; Montalbano, A.; Barraja, P.; Diana, P.; Cirrincione, G.; Cascioferro, S. Pharmaceutical Approaches to Target Antibiotic Resistance Mechanisms. J. Med. Chem. 2017, 60, 8268–8297. [Google Scholar] [CrossRef]
- Lamut, A.; Peterlin Mašič, L.; Kikelj, D.; Tomašič, T. Efflux pump inhibitors of clinically relevant multidrug resistant bacteria. Med. Res. Rev. 2019, 39, 2460–2504. [Google Scholar] [CrossRef]
- Martinez, J.L.; Sánchez, M.B.; Martínez-Solano, L.; Hernandez, A.; Garmendia, L.; Fajardo, A.; Alvarez-Ortega, C. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 2009, 33, 430–449. [Google Scholar] [CrossRef]
- Kaatz, G.W.; Seo, S.M.; Ruble, C.A. Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1993, 37, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Kaatz, G.W.; Seo, S.M.; Ruble, C.A. Mechanisms of Fluoroquinolone Resistance in Staphylococcus aureus. J. Infect. Dis. 1991, 163, 1080–1086. [Google Scholar] [CrossRef]
- Novick, R. Properties of a cryptic high-frequency transducing phage in Staphylococcus aureus. Virology 1967, 33, 155–166. [Google Scholar] [CrossRef]
- Price, C.T.D.; Kaatz, G.W.; Gustafson, J.E. The multidrug efflux pump NorA is not required for salicylate-induced reduction in drug accumulation by Staphylococcus aureus. Int. J. Antimicrob. Agents 2002, 20, 206–213. [Google Scholar] [CrossRef]
- Tegos, G.; Stermitz, F.R.; Lomovskaya, O.; Lewis, K. Multidrug Pump Inhibitors Uncover Remarkable Activity of Plant Antimicrobials. Antimicrob. Agents Chemother. 2002, 46, 3133. [Google Scholar] [CrossRef] [Green Version]
- Kaatz, G.W.; Seo, S.M. Inducible NorA-Mediated Multidrug Resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1995, 39, 2650–2655. [Google Scholar] [CrossRef] [Green Version]
- Gambino, L.; Gracheck, S.J.; Miller, P.F. Overexpression of the MarA positive regulator is sufficient to confer multiple antibiotic resistance in Escherichia coli. J. Bacteriol. 1993, 175, 2888–2894. [Google Scholar] [CrossRef] [Green Version]
- Mallea, M.; Chevalier, J.; Bornet, C.; Eyraud, A.; Davin-Regli, A.; Bollet, C.; Pages, J.M. Porin alteration and active efflux: Two in vivo drug resistance strategies used by Enterobacter aerogenes. Microbiology 1998, 144, 3003–3009. [Google Scholar] [CrossRef] [Green Version]
- Ghisalberti, D.; Masi, M.; Pagès, J.M.; Chevalier, J. Chloramphenicol and expression of multidrug efflux pump in Enterobacter aerogenes. Biochem. Biophys. Res. Commun. 2005, 328, 1113–1118. [Google Scholar] [CrossRef]
- Pradel, E.; Pagès, J.M. The AcrAB–TolC efflux pump contributes to multidrug resistance in the nosocomial pathogen Enterobacter aerogenes. Antimicrob. Agents Chemother. 2002, 46, 2640–2643. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Fukuda, Y.; Murata, K.; Kimura, A. Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 1983, 153, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viveiros, M.; Jesus, A.; Brito, M.; Leandro, C.; Martins, M.; Ordway, D.; Molnar, A.M.; Molnar, J.; Amaral, L. Inducement and Reversal of Tetracycline Resistance in Escherichia coli K-12 and Expression of Proton Gradient-Dependent Multidrug Efflux Pump Genes. Antimicrob. Agents Chemother. 2005, 49, 3578. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Wang, H.; Zou, L.; Zhang, A.; Yang, X.; Guan, Z. Evaluation and Target Validation of Indole Derivatives as Inhibitors of the AcrAB–TolC Efflux Pump. Biosci. Biotechnol. Biochem. 2010, 74, 2237–2241. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, J.; Pagès, J.M.; Eyraud, A.; Malléa, M. Membrane Permeability Modifications Are Involved in Antibiotic Resistance in Klebsiella pneumoniae. Biochem. Biophys. Res. Commun. 2000, 274, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Snapper, S.B.; Melton, R.E.; Mustafa, S.; Kieser, T.; Jacobs, W.R. Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 1990, 4, 1911–1919. [Google Scholar] [CrossRef]
- Horan, K.L.; Freeman, R.; Weigel, K.; Semret, M.; Pfaller, S.; Covert, T.C.; van Soolingen, D.; Leao, S.C.; Behr, M.A.; Cangelosi, G.A. Isolation of the genome sequence strain Mycobacterium avium 104 from multiple patients over a 17-year period. J. Clin. Microbiol. 2006, 44, 783–789. [Google Scholar] [CrossRef] [Green Version]
- Machado, D.; Cannalire, R.; Santos Costa, S.; Manfroni, G.; Tabarrini, O.; Cecchetti, V.; Couto, I.; Viveiros, M.; Sabatini, S. Boosting Effect of 2-Phenylquinoline Efflux Inhibitors in Combination with Macrolides against Mycobacterium smegmatis and Mycobacterium avium. ACS Infect. Dis. 2016, 1, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug Efflux Pumps in Staphylococcus aureus: An Update. Open Microbiol. J. 2013, 7, 59–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S. Multidrug efflux pumps in Staphylococcus aureus and their clinical implications. J. Microbiol. 2016, 54, 1–8. [Google Scholar] [CrossRef]
- Ubukata, K.; Itoh-Yamashita, N.; Konno, M. Cloning and expression of the norA gene for fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1989, 33, 1535–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Bogaki, M.; Nakamura, S.; Ubukata, K.; Konno, M. Nucleotide sequence and characterization of the Staphylococcus aureus norA gene, which confers resistance to quinolones. J. Bacteriol. 1990, 172, 6942–6949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskar, B.V.; Chandra Babu, T.M.; Reddy, N.V.; Rajendra, W. Homology modeling, molecular dynamics, and virtual screening of NorA efflux pump inhibitors of Staphylococcus aureus. Drug Des. Devel. Ther. 2016, 10, 3237–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tintino, S.R.; de Souza, V.C.A.; da Silva, J.M.A.; Oliveira-Tintino, C.D.; Pereira, P.S.; Leal-Balbino, T.C.; Pereira-Neves, A.; Siqueira-Junior, J.P.; da Costa, J.G.M.; Rodrigues, F.F.G.; et al. Effect of Vitamin K3 Inhibiting the Function of NorA Efflux Pump and Its Gene Expression on Staphylococcus aureus. Membranes 2020, 10, 130. [Google Scholar] [CrossRef] [PubMed]
- Palazzotti, D.; Bissaro, M.; Bolcato, G.; Astolfi, A.; Felicetti, T.; Sabatini, S.; Sturlese, M.; Cecchetti, V.; Barreca, M.L.; Moro, S. Deciphering the Molecular Recognition Mechanism of Multidrug Resistance Staphylococcus aureus NorA Efflux Pump Using a Supervised Molecular Dynamics Approach. Int. J. Mol. Sci. 2019, 20, 4041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, P.N.; Westhaus, E.; Klyachko, K.; Johnson, M.E.; Neyfakh, A.A. Multiple novel inhibitors of the NorA multidrug transporter of Staphylococcus aureus. Antimicrob. Agents Chemother. 1999, 43, 2404–2408. [Google Scholar] [CrossRef] [Green Version]
- Neyfakh, A.A.; Borsch, C.M.; Kaatz, G.W. Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob. Agents Chemother. 1993, 37, 128–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Lyass, L.; Markham, P.N.; Taylor, S.S.; Vazquez-Laslop, N.; Neyfakh, A.A. Two highly similar multidrug transporters of Bacillus subtilis whose expression is differentially regulated. J. Bacteriol. 1995, 177, 3904–3910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samosorn, S.; Bremner, J.; Ball, A.; Lewis, K. Synthesis of functionalized 2-aryl-5-nitro-1H-indoles and their activity as bacterial NorA efflux pump inhibitors. Bioorg. Med. Chem. 2006, 14, 857–865. [Google Scholar] [CrossRef] [PubMed]
- dit Chabert, J.F.; Marquez, B.; Neville, L.; Joucla, L.; Broussous, S.; Bouhours, P.; David, E.; Pellet-Rostaing, S.; Marquet, B.; Moreau, N.; et al. Synthesis and evaluation of new arylbenzo[b]thiophene and diarylthiophene derivatives as inhibitors of the NorA multidrug transporter of Staphylococcus aureus. Bioorg. Med. Chem. 2007, 15, 4482–4497. [Google Scholar] [CrossRef]
- Ambrus, J.I.; Kelso, M.J.; Bremner, J.B.; Ball, A.R.; Casadei, G.; Lewis, K. Structure-activity relationships of 2-aryl-1H-indole inhibitors of the NorA efflux pump in Staphylococcus aureus. Bioorg. Med. Chem. Lett. 2008, 18, 4294–4297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hequet, A.; Burchak, O.N.; Jeanty, M.; Guinchard, X.; Le Pihive, E.; Maigre, L.; Bouhours, P.; Schneider, D.; Maurin, M.; Paris, J.M.; et al. 1-(1H-indol-3-yl)ethanamine derivatives as potent Staphylococcus aureus NorA efflux pump inhibitors. ChemMedChem 2014, 9, 1534–1545. [Google Scholar] [CrossRef]
- Burchak, O.N.; Le Pihive, E.; Maigre, L.; Guinchard, X.; Bouhours, P.; Jolivalt, C.; Schneider, D.; Maurin, M.; Giglione, C.; Meinnel, T.; et al. Synthesis and evaluation of 1-(1H-indol-3-yl)ethanamine derivatives as new antibacterial agents. Bioorg. Med. Chem. 2011, 19, 3204–3215. [Google Scholar] [CrossRef]
- Liger, F.; Pellet-Rostaing, S.; Popowycz, F.; Lemaire, M. Bromination of 3-substituted benzo[b]thiophenes: Access to Raloxifen intermediate. Tetrahedron Lett. 2011, 52, 3736–3739. [Google Scholar] [CrossRef]
- Liger, F.; Bouhours, P.; Ganem-Elbaz, C.; Jolivalt, C.; Pellet-Rostaing, S.; Popowycz, F.; Paris, J.-M.; Lemaire, M. C2 Arylated Benzo[b]thiophene Derivatives as Staphylococcus aureus NorA Efflux Pump Inhibitors. ChemMedChem 2016, 11, 320–330. [Google Scholar] [CrossRef]
- Lepri, S.; Buonerba, F.; Goracci, L.; Velilla, I.; Ruzziconi, R.; Schindler, B.D.; Seo, S.M.; Kaatz, G.W.; Cruciani, G. Indole Based Weapons to Fight Antibiotic Resistance: A Structure-Activity Relationship Study. J. Med. Chem. 2016, 59, 867–891. [Google Scholar] [CrossRef] [PubMed]
- Baroni, M.; Cruciani, G.; Sciabola, S.; Perruccio, F.; Mason, J.S. A Common Reference Framework for Analyzing/Comparing Proteins and Ligands. Fingerprints for Ligands And Proteins (FLAP): Theory and Application. J. Chem. Inf. Model. 2007, 47, 279–294. [Google Scholar] [CrossRef]
- Sabatini, S.; Gosetto, F.; Iraci, N.; Barreca, M.L.; Massari, S.; Sancineto, L.; Manfroni, G.; Tabarrini, O.; Dimovska, M.; Kaatz, G.W.; et al. Re-evolution of the 2-phenylquinolines: Ligand-based design, synthesis, and biological evaluation of a potent new class of Staphylococcus aureus NorA efflux pump inhibitors to combat antimicrobial resistance. J. Med. Chem. 2013, 56, 4975–4989. [Google Scholar] [CrossRef]
- Buonerba, F.; Lepri, S.; Goracci, L.; Schindler, B.D.; Seo, S.M.; Kaatz, G.W.; Cruciani, G. Improved Potency of Indole-Based NorA Efflux Pump Inhibitors: From Serendipity toward Rational Design and Development. J. Med. Chem. 2017, 60, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Schindler, B.D.; Jacinto, P.; Kaatz, G.W. Inhibition of drug efflux pumps in Staphylococcus aureus: Current status of potentiating existing antibiotics. Future Microbiol. 2013, 8, 491–507. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, S.; Gosetto, F.; Manfroni, G.; Tabarrini, O.; Kaatz, G.W.; Patel, D.; Cecchetti, V. Evolution from a Natural Flavones Nucleus to Obtain 2-(4-Propoxyphenyl)quinoline Derivatives As Potent Inhibitors of the S. aureus NorA Efflux Pump. J. Med. Chem. 2011, 54, 5722–5736. [Google Scholar] [CrossRef] [PubMed]
- Zloh, M.; Gibbons, S. Molecular Similarity of MDR Inhibitors. Int. J. Mol. Sci 2004, 5, 37–47. [Google Scholar] [CrossRef]
- Felicetti, T.; Cannalire, R.; Pietrella, D.; Latacz, G.; Lubelska, A.; Manfroni, G.; Barreca, M.L.; Massari, S.; Tabarrini, O.; Kieć-Kononowicz, K.; et al. 2-Phenylquinoline S. aureus NorA efflux pump inhibitors: Evaluation of the importance of methoxy group introduction. J. Med. Chem. 2018, 61, 7827–7848. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, T.; Cannalire, R.; Nizi, M.G.; Tabarrini, O.; Massari, S.; Barreca, M.L.; Manfroni, G.; Schindler, B.D.; Cecchetti, V.; Kaatz, G.W.; et al. Studies on 2-phenylquinoline Staphylococcus aureus NorA efflux pump inhibitors: New insights on the C-6 position. Eur. J. Med. Chem. 2018, 155, 428–433. [Google Scholar] [CrossRef]
- Felicetti, T.; Mangiaterra, G.; Cannalire, R.; Cedraro, N.; Pietrella, D.; Astolfi, A.; Massari, S.; Tabarrini, O.; Manfroni, G.; Barreca, M.L.; et al. C-2 phenyl replacements to obtain potent quinoline-based Staphylococcus aureus NorA inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Cannalire, R.; Mangiaterra, G.; Felicetti, T.; Astolfi, A.; Cedraro, N.; Massari, S.; Manfroni, G.; Tabarrini, O.; Vaiasicca, S.; Barreca, M.L.; et al. Structural Modifications of the Quinolin-4-yloxy Core to Obtain New Staphylococcus aureus NorA Inhibitors. Int. J. Mol. Sci. 2020, 21, 7037. [Google Scholar] [CrossRef]
- Cedraro, N.; Cannalire, R.; Astolfi, A.; Mangiaterra, G.; Felicetti, T.; Vaiasicca, S.; Cernicchi, G.; Massari, S.; Manfroni, G.; Tabarrini, O.; et al. From Quinoline to Quinazoline-Based S. aureus NorA Efflux Pump Inhibitors by Coupling a Focused Scaffold Hopping Approach and a Pharmacophore Search. ChemMedChem 2021, 16, 3044–3059. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Felicetti, T.; Iraci, N.; Manfroni, G.; Massari, S.; Pietrella, D.; Tabarrini, O.; Kaatz, G.W.; Barreca, M.L.; Sabatini, S.; et al. Pharmacophore-based repositioning of approved drugs as novel Staphylococcus aureus NorA efflux pump inhibitors. J. Med. Chem. 2017, 60, 1598–1604. [Google Scholar] [CrossRef] [PubMed]
- Silhavy, T.J.; Kahne, D.; Walker, S. The Bacterial Cell Envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000414. [Google Scholar] [CrossRef] [PubMed]
- Delcour, A.H. Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta 2009, 1794, 808. [Google Scholar] [CrossRef] [Green Version]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 2002, 419, 587–593. [Google Scholar] [CrossRef]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Hayashi, K.; Nagata, C.; Hoshino, K.; Onodera, Y.; Nishino, K.; Yamaguchi, A. Structural basis for the inhibition of bacterial multidrug exporters. Nature 2013, 500, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Richmond, G.E.; Piddock, V.L.J. Multidrug efflux pumps in Gram-negative bacteria and their role in antibiotic resistance. Future Microbiol. 2014, 9, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Kwasny, S.M.; Kim, H.S.; Nguyen, S.T.; Houseweart, C.; D’Souza, S.; Walker, G.C.; Peet, N.P.; Nikaido, H.; Bowlin, T.L. Characterization of a novel pyranopyridine inhibitor of the AcrAB efflux pump of Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargiu, A.V.; Ruggerone, P.; Opperman, T.J.; Nguyen, S.T.; Nikaido, H. Molecular mechanism of MBX2319 inhibition of Escherichia coli AcrB multidrug efflux pump and comparison with other inhibitors. Antimicrob. Agents Chemother. 2014, 58, 6224–6234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.T.; Kwasny, S.M.; Ding, X.; Cardinale, S.C.; McCarthy, C.T.; Kim, H.-S.; Nikaido, H.; Peet, N.P.; Williams, J.D.; Bowlin, T.L.; et al. Structure–activity relationships of a novel pyranopyridine series of Gram-negative bacterial efflux pump inhibitors. Bioorg. Med. Chem. 2015, 23, 2024–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron, Z.; Opperman, T.J.; Pucci, M.J.; Dougherty, T.J. Optimization of a novel series of pyranopyridine RND efflux pump inhibitors. Curr. Opin. Microbiol. 2016, 33, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Sjuts, H.; Vargiu, A.V.; Kwasny, S.M.; Nguyen, S.T.; Kim, H.-S.; Ding, X.; Ornik, A.R.; Ruggerone, P.; Bowlin, T.L.; Nikaido, H.; et al. Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, 3509–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, A.; Nakashima, R.; Sakurai, K. Structural basis of RND-type multidrug exporters. Front. Microbiol. 2015, 6, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevalier, J.; Atifi, S.; Eyraud, A.; Mahamoud, A.; Barbe, J.; Pagès, J.M. New pyridoquinoline derivatives as potential inhibitors of the fluoroquinolone efflux pump in resistant Enterobacter aerogenes strains. J. Med. Chem. 2001, 44, 4023–4026. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Regulation of chromosomally mediated multiple antibiotic resistance: The mar regulon. Antimicrob. Agents Chemother. 1997, 41, 2067–2075. [Google Scholar] [CrossRef] [Green Version]
- Keeney, D.; Ruzin, A.; McAleese, F.; Murphy, E.; Bradford, P.A. MarA-mediated overexpression of the AcrAB efflux pump results in decreased susceptibility to tigecycline in Escherichia coli. J. Antimicrob. Chemother. 2008, 61, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Gallo, S.; Chevalier, J.; Mahamoud, A.; Eyraud, A.; Pagès, J.M.; Barbe, J. 4-alkoxy and 4-thioalkoxyquinoline derivatives as chemosensitizers for the chloramphenicol-resistant clinical Enterobacter aerogenes 27 strain. Int. J. Antimicrob. Agents 2003, 22, 270–273. [Google Scholar] [CrossRef]
- Malléa, M.; Mahamoud, A.; Chevalier, J.; Alibert-Franco, S.; Brouant, P.; Barbe, J.; Pages, J.-M. Alkylaminoquinolines inhibit the bacterial antibiotic efflux pump in multidrug-resistant clinical isolates. Biochem. J. 2003, 376, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Lamers, R.P.; Cavallari, J.F.; Burrows, L.L. The Efflux Inhibitor Phenylalanine-Arginine Beta-Naphthylamide (PAβN) Permeabilizes the Outer Membrane of Gram-Negative Bacteria. PLoS ONE 2013, 8, e60666. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, J.; Bredin, J.; Mahamoud, A.; Malléa, M.; Barbe, J.; Pagès, J.M. Inhibitors of Antibiotic Efflux in Resistant Enterobacter aerogenes and Klebsiella pneumoniae Strains. Antimicrob. Agents Chemother. 2004, 48, 1043–1046. [Google Scholar] [CrossRef] [Green Version]
- Ghisalberti, D.; Mahamoud, A.; Chevalier, J.; Baitiche, M.; Martino, M.; Pagès, J.M.; Barbe, J. Chloroquinolines block antibiotic efflux pumps in antibiotic-resistant Enterobacter aerogenes isolates. Int. J. Antimicrob. Agents 2006, 27, 565–569. [Google Scholar] [CrossRef]
- Machado, D.; Fernandes, L.; Costa, S.S.; Cannalire, R.; Manfroni, G.; Tabarrini, O.; Couto, I.; Sabatini, S.; Viveiros, M. Mode of action of the 2-phenylquinoline efflux inhibitor PQQ4R against Escherichia coli. PeerJ 2017, 5, e3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadelis, M.M.; Li, S.A.; Bourguet-Kondracki, M.-L.; Blanchet, M.; Douafer, H.; Brunel, J.M.; Copp, B.R. Spermine Derivatives of Indole-3-carboxylic Acid, Indole-3-acetic Acid and Indole-3-acrylic Acid as Gram-Negative Antibiotic Adjuvants. ChemMedChem 2021, 16, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Pang, Y.; Wang, Y.; Cohen, C.; Zhao, Y.; Liu, C. Differences in risk factors and drug susceptibility between Mycobacterium avium and Mycobacterium intracellulare lung diseases in China. Int. J. Antimicrob. Agents 2015, 45, 491–495. [Google Scholar] [CrossRef]
- Jones, M.M.; Winthrop, K.L.; Nelson, S.D.; Duvall, S.L.; Patterson, O.V.; Nechodom, K.E.; Findley, K.E.; Radonovich, L.J., Jr.; Samore, M.H.; Fennelly, K.P. Epidemiology of nontuberculous mycobacterial infections in the U.S. Veterans Health Administration. PLoS ONE 2018, 13, e0197976. [Google Scholar] [CrossRef] [PubMed]
- Schmalstieg, A.M.; Srivastava, S.; Belkaya, S.; Deshpande, D.; Meek, C.; Leff, R.; Van Oers, N.S.C.; Gumbo, T. The antibiotic resistance arrow of time: Efflux pump induction is a general first step in the evolution of mycobacterial drug resistance. Antimicrob. Agents Chemother. 2012, 56, 4806–4815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, L.; Sampaio, D.; Couto, I.; Machado, D.; Kern, W.V.; Amaral, L.; Viveiros, M. The role of efflux pumps in macrolide resistance in Mycobacterium avium complex. Int. J. Antimicrob. Agents 2009, 34, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.; Ramos, J.; Couto, I.; Amaral, L.; Viveiros, M. Ethidium bromide transport across Mycobacterium smegmatis cell-wall: Correlation with antibiotic resistance. BMC Microbiol. 2011, 11, 35. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.; Wagner, D.; Viveiros, M.; Sampaio, D.; Couto, I.; Vavra, M.; Kern, W.V.; Amaral, L. Thioridazine and chlorpromazine inhibition of ethidium bromide efflux in Mycobacterium avium and Mycobacterium smegmatis. J. Antimicrob. Chemother. 2008, 61, 1076–1082. [Google Scholar] [CrossRef]
- Lechner, D.; Gibbons, S.; Bucar, F. Plant phenolic compounds as ethidium bromide efflux inhibitors in Mycobacterium smegmatis. J. Antimicrob. Chemother. 2008, 62, 345–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rindi, L. Efflux Pump Inhibitors against Nontuberculous Mycobacteria. Int. J. Mol. Sci. 2020, 21, 4191. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Machado, D.; Felicetti, T.; Santos Costa, S.; Massari, S.; Manfroni, G.; Barreca, M.L.; Tabarrini, O.; Couto, I.; Viveiros, M.; et al. Natural isoflavone biochanin A as a template for the design of new and potent 3-phenylquinolone efflux inhibitors against Mycobacterium avium. Eur. J. Med. Chem. 2017, 140, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Felicetti, T.; Machado, D.; Cannalire, R.; Astolfi, A.; Massari, S.; Tabarrini, O.; Manfroni, G.; Barreca, M.L.; Cecchetti, V.; Viveiros, M.; et al. Modifications on C6 and C7 Positions of 3-Phenylquinolone Efflux Pump Inhibitors Led to Potent and Safe Antimycobacterial Treatment Adjuvants. ACS Infect. Dis. 2019, 5, 982–1000. [Google Scholar] [CrossRef]
- Yoshida, K.-I.; Nakayama, K.; Ohtsuka, M.; Kuru, N.; Yokomizo, Y.; Sakamoto, A.; Takemura, M.; Hoshino, K.; Kanda, H.; Nitanai, H.; et al. MexAB-OprM specific efflux pump inhibitors in Pseudomonas aeruginosa. Part 7: Highly soluble and in vivo active quaternary ammonium analogue D13-9001, a potential preclinical candidate. Bioorg. Med. Chem. 2007, 15, 7087–7097. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fan, G.; Hryc, C.F.; Blaza, J.N.; Serysheva, I.I.; Schmid, M.F.; Chiu, W.; Luisi, B.F.; Du, D. An allosteric transport mechanism for the AcrAB–TolC multidrug efflux pump. Elife 2017, 6, e24905. [Google Scholar] [CrossRef]
- Glavier, M.; Puvanendran, D.; Salvador, D.; Decossas, M.; Phan, G.; Garnier, C.; Frezza, E.; Cece, Q.; Schoehn, G.; Picard, M.; et al. Antibiotic export by MexB multidrug efflux transporter is allosterically controlled by a MexA-OprM chaperone-like complex. Nat. Commun. 2020, 11, 4948. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strains | Description | Ref. |
|---|---|---|
| B. subtilis | ||
| ΔΔ | genes that encode the multidrug transporters Bmr and Blt are genetically inactivated | [26] |
| ΔΔNA | expresses a functional NorA transporter from the plasmid expression vector pBEV | [27] |
| S. aureus | ||
| SA-1199 | wild-type | [27] |
| SA-1199B | overexpresses the chromosomal norA gene which harbors a mutation in grlA | [28] |
| 8325-4 | wild-type | [29] |
| K1758 | ΔnorA | [30] |
| K2361 | SA-K1758 with pK364:norA | [31] |
| ATCC 25923 | wild-type | |
| SA-K2378 | having a plasmid that results in overexpression of norA from S. aureus SA1199 | [32] |
| SA-K1902 | ΔnorA | [32] |
| E. aerogenes | ||
| ATCC 13048 | wild-type | |
| ATCC 13048 p9 | overexpressing the EP activator marA gene | [33] |
| EA27 | overexpressing EP, clinically isolated | [34] |
| EA3 | overexpressing EP, clinically isolated | [34] |
| EA5 | overexpressing EP, clinically isolated | [34] |
| CM-64 | resistant to CAF due to EP overexpressing, | [35] |
| EA117 | low porin levels and also substitutions in the QRDR domain of GrlA | [36] |
| EA289 | overexpressing AcrAB–TolC pump | [36] |
| E. coli | ||
| AG100 | wild-type | |
| AG100A | AcrAB pump-deficient | [37] |
| AG100tet | overexpressing efflux pump | [38] |
| ATCC 25922 | wild-type | |
| YD02 | multidrug-resistant after induction from ATCC 25922 | [39] |
| P. aeruginosa | ||
| ATCC 27853 | wild-type | |
| K. pneumoniae | ||
| KP55 | porin deficient phenotype | [40] |
| M. smegmatis mc2155 | ||
| ATCC 700084 | wild-type | [41] |
| M. avium | ||
| 104 | wild-type | [42] |
| 104CLA3 | resistant to CLA due to overexpression of efflux pumps | [43] |
| 104CLA4 | resistant to CLA due to overexpressing efflux pumps and harboring the mutation A-2059G in 23S rRNA | [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cernicchi, G.; Felicetti, T.; Sabatini, S. Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives. Molecules 2021, 26, 6996. https://doi.org/10.3390/molecules26226996
Cernicchi G, Felicetti T, Sabatini S. Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives. Molecules. 2021; 26(22):6996. https://doi.org/10.3390/molecules26226996
Chicago/Turabian StyleCernicchi, Giada, Tommaso Felicetti, and Stefano Sabatini. 2021. "Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives" Molecules 26, no. 22: 6996. https://doi.org/10.3390/molecules26226996
APA StyleCernicchi, G., Felicetti, T., & Sabatini, S. (2021). Microbial Efflux Pump Inhibitors: A Journey around Quinoline and Indole Derivatives. Molecules, 26(22), 6996. https://doi.org/10.3390/molecules26226996