
Drug Design: Where We Are and Future Prospects


Abstract
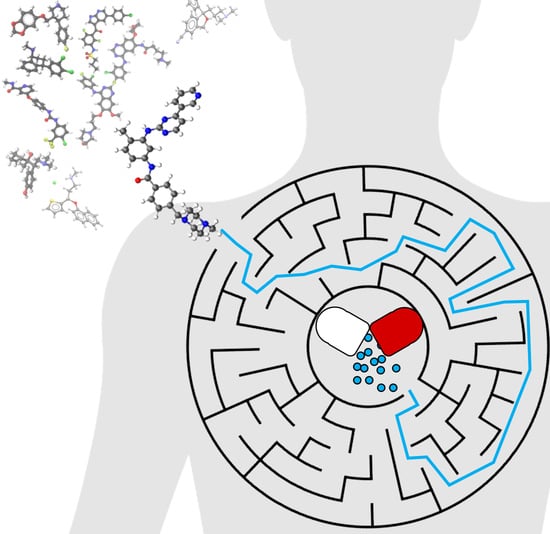
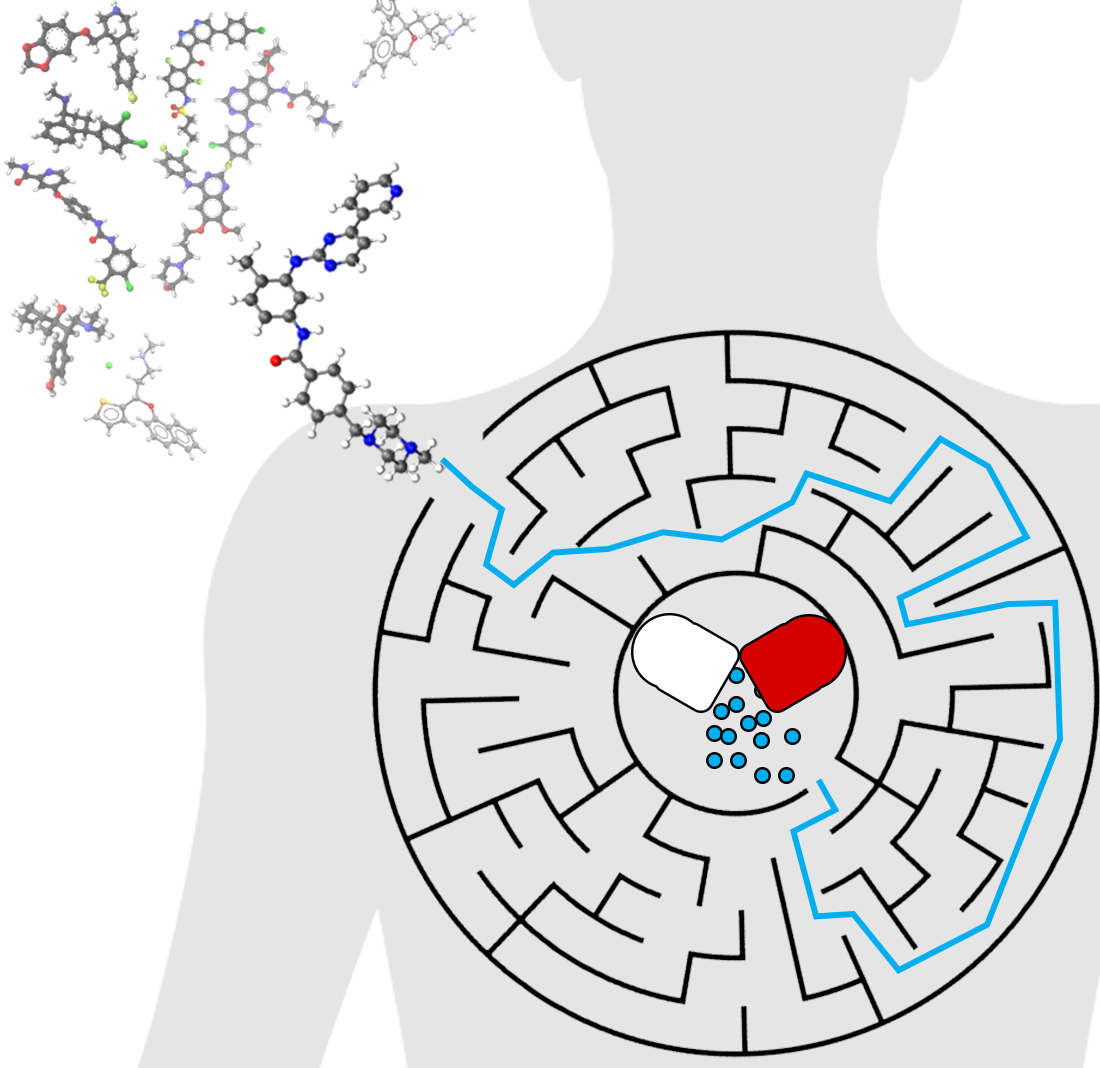
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Lead Discovery
2.1. Target Selection and Validation: Possible Expansion of Chemical Space
2.2. From Hit to Lead: Structure-Guided Drug Design and Beyond
2.3. Speeding up Screening and Design: Artificial Intelligence in Drug Discovery
2.4. One Size Does Not Fit All: From General to Precision Medicine
3. Pharmacokinetics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Druchok, M.; Yarish, D.; Gurbych, O.; Maksymenko, M. Toward efficient generation, correction, and properties control of unique drug-like structures. J. Comput. Chem. 2021, 42, 746–760. [Google Scholar] [CrossRef]
- Yang, W.; Gadgil, P.; Krishnamurthy, V.R.; Landis, M.; Mallick, P.; Patel, D.; Patel, P.J.; Reid, D.L.; Sanchez-Felix, M. The Evolving Druggability and Developability Space: Chemically Modified New Modalities and Emerging Small Molecules. AAPS J. 2020, 22, 21. [Google Scholar] [CrossRef]
- Roses, A.D.; Burns, D.K.; Chissoe, S.; Middleton, L.; Jean, P.S. Keynote review: Disease-specific target selection: A critical first step down the right road. Drug Discov. Today 2005, 10, 177–189. [Google Scholar] [CrossRef]
- Steadman, V.A. Drug Discovery: Collaborations between Contract Research Organizations and the Pharmaceutical Industry. ACS Med. Chem. Lett. 2018, 9, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decorte, B.L. Evolving Outsourcing Landscape in Pharma R&D: Different Collaborative Models and Factors to Consider When Choosing a Contract Research Organization. J. Med. Chem. 2020, 63, 11362–11367. [Google Scholar] [CrossRef]
- Hingorani, A.D.; Kuan, V.; Finan, C.; Kruger, F.A.; Gaulton, A.; Chopade, S.; Sofat, R.; MacAllister, R.J.; Overington, J.P.; Hemingway, H.; et al. Improving the odds of drug development success through human genomics: Modelling study. Sci. Rep. 2019, 9, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, J.; Gromo, G. Target selection in drug discovery. Nat. Rev. Drug Discov. 2003, 2, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Dugger, S.A.; Platt, A.; Goldstein, D.B. Drug development in the era of precision medicine. Nat. Rev. Drug Discov. 2018, 17, 183–196. [Google Scholar] [CrossRef]
- Boyle, E.A.; Li, Y.I.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
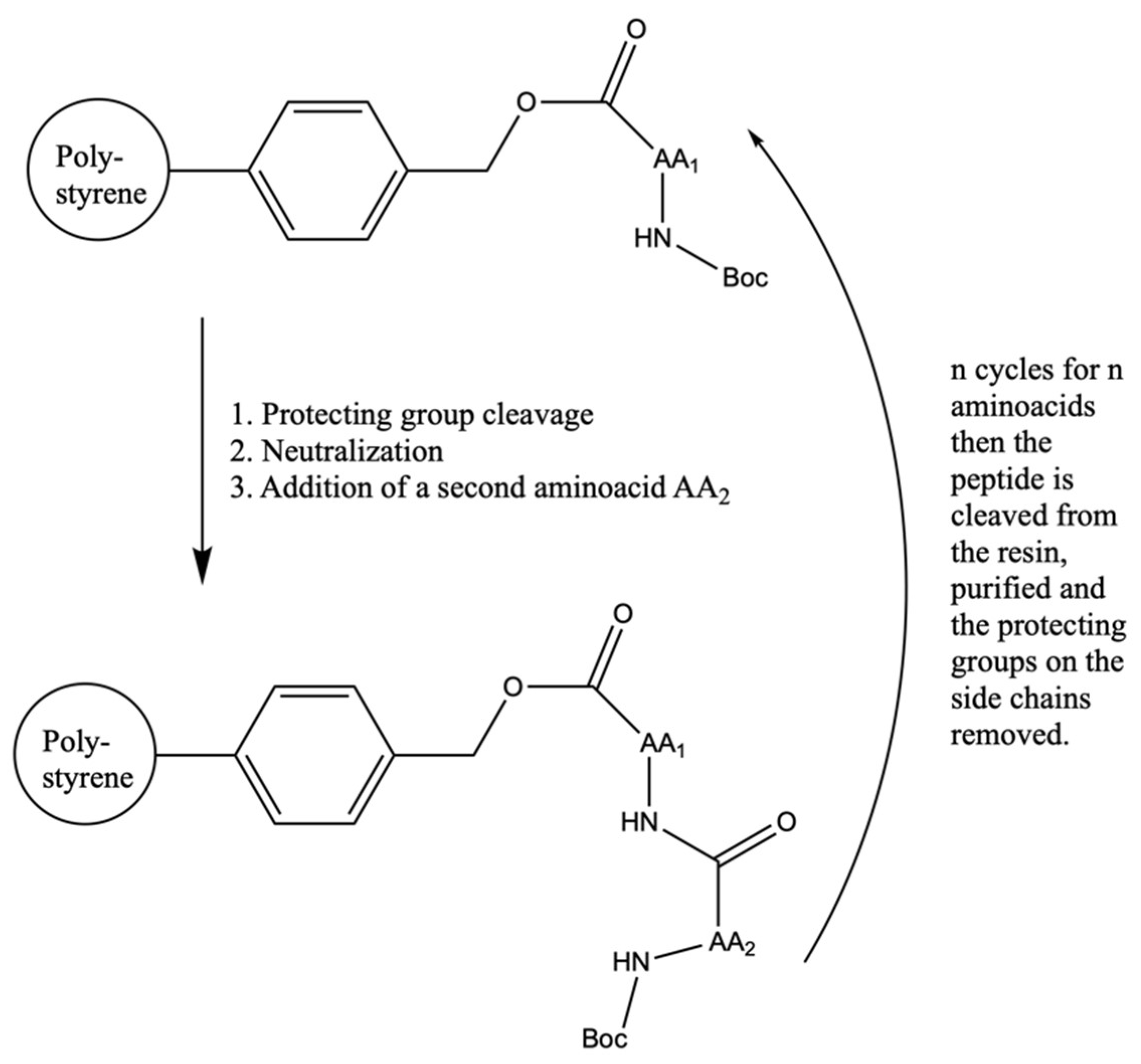
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
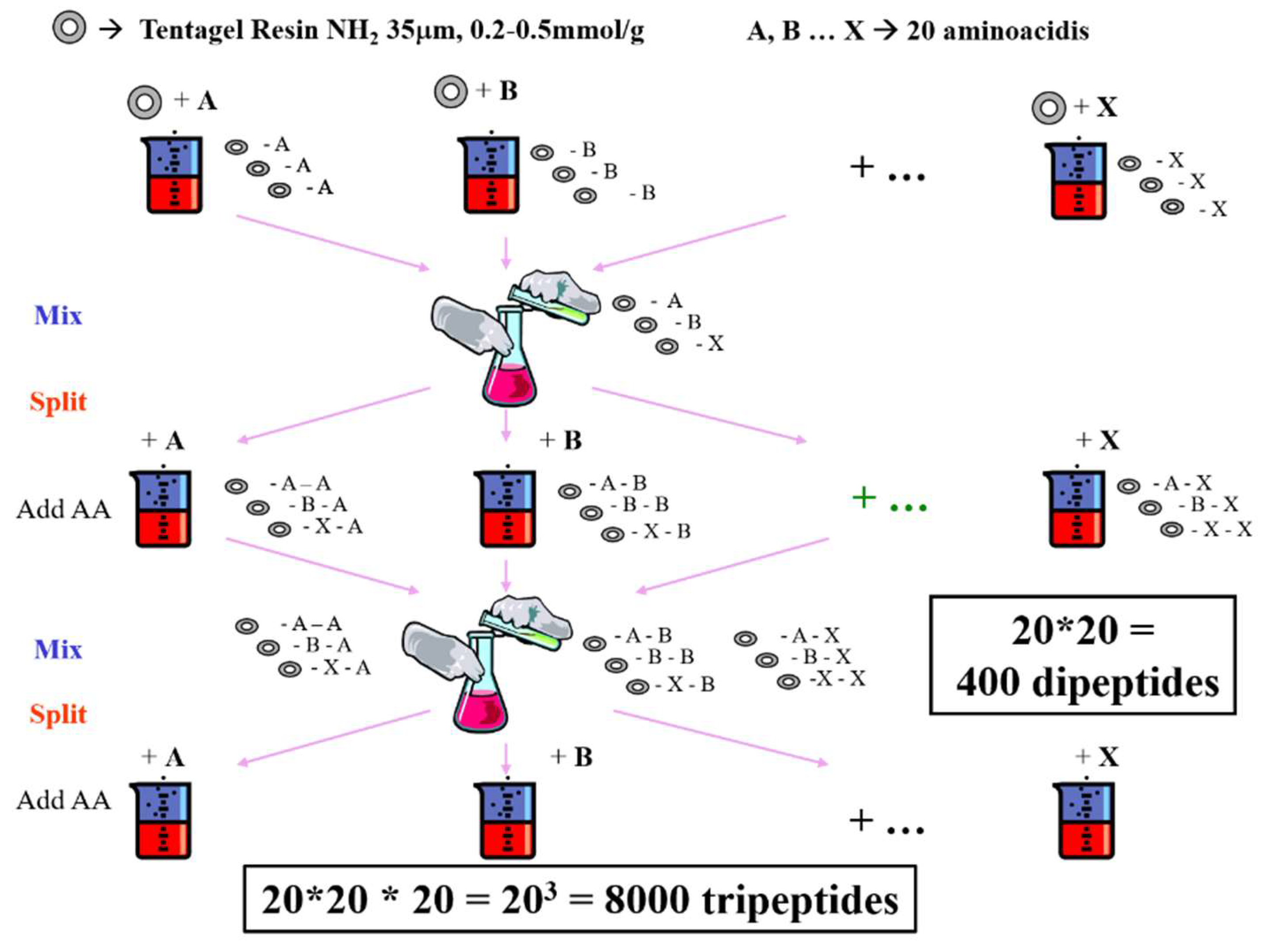
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmeierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, M.D.; Caruthers, M.H. Synthesis of Deoxyoligonucleotides on a Polymer Support. J. Am. Chem. Soc. 1981, 103, 3185–3191. [Google Scholar] [CrossRef]
- Marshall, W.S.; Boymel, J.L. Oligonucleotide synthesis as a tool in drug discovery research. Drug Discov. Today 1998, 3, 34–42. [Google Scholar] [CrossRef]
- Bunin, B.A.; Ellman, J.A. A General and Expedient Method for the Solid-Phase Synthesis of 1,4-Benzodiazepine Derivatives. J. Am. Chem. Soc. 1992, 114, 10997–10998. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Xiao, X.-Y.; Parandoosh, Z.; Senyei, A.; Nova, M.P. Radiofrequency Encoded Combinatorial Chemistry. Angew. Chem. Int. Ed. Engl. 1995, 34, 2289–2291. [Google Scholar] [CrossRef]
- Seeberger, P.H.; Haase, W.C. Solid-phase oligosaccharide synthesis and combinatorial carbohydrate libraries. Chem. Rev. 2000, 100, 4349–4393. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohkawa, Y.; Yamauchi, Y.; Hamamura, K.; Ohmi, Y.; Furukawa, K. Fine tuning of cell signals by glycosylation. J. Biochem. 2012, 151, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Seeberger, P.H. Discovery of Semi- and Fully-Synthetic Carbohydrate Vaccines against Bacterial Infections Using a Medicinal Chemistry Approach. Chem. Rev. 2021, 121, 3598–3626. [Google Scholar] [CrossRef]
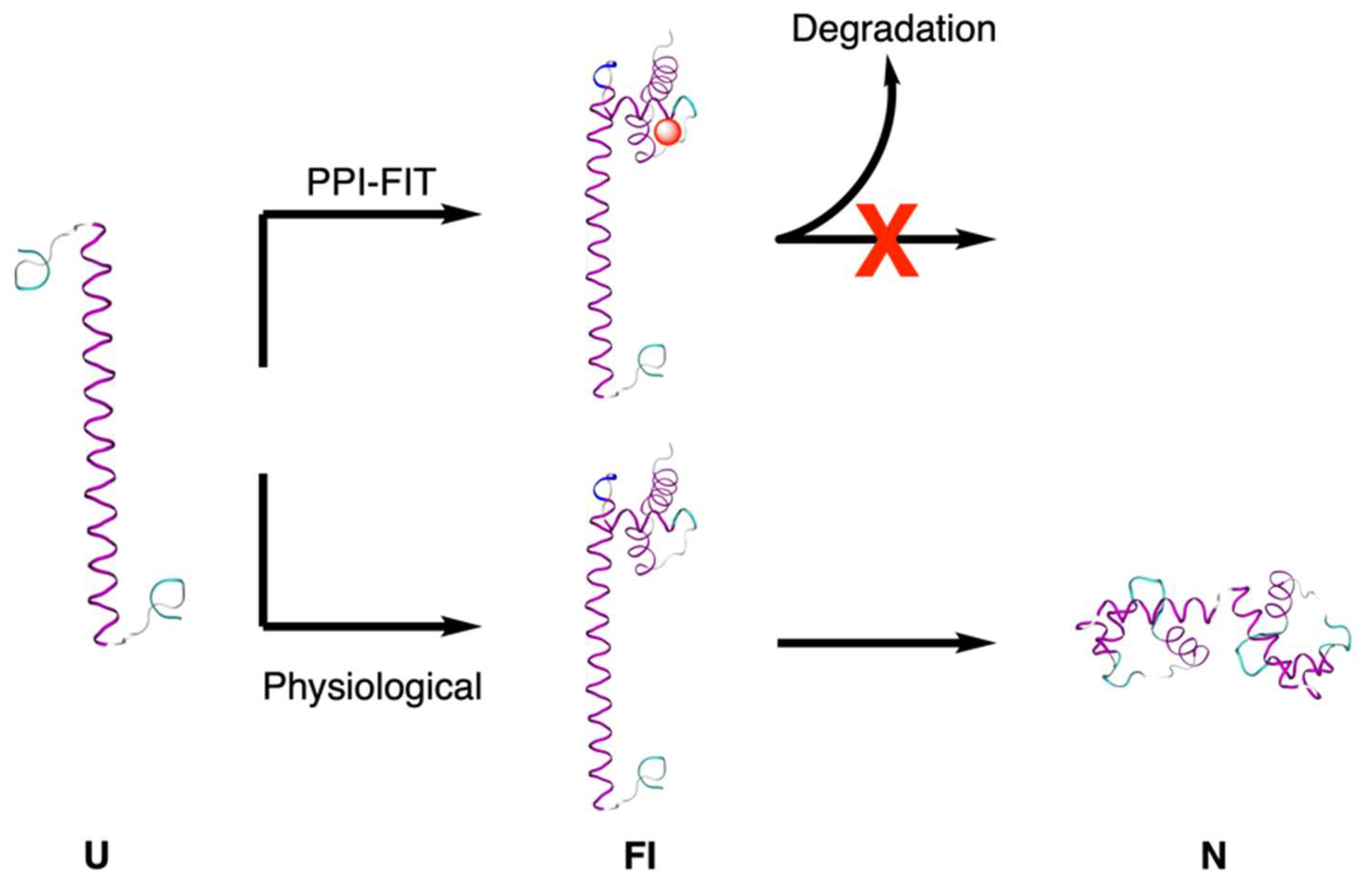
- Spagnolli, G.; Massignan, T.; Astolfi, A.; Biggi, S.; Rigoli, M.; Brunelli, P.; Libergoli, M.; Ianeselli, A.; Orioli, S.; Boldrini, A.; et al. Pharmacological inactivation of the prion protein by targeting a folding intermediate. Commun. Biol. 2021, 4, 1–16. [Google Scholar] [CrossRef]
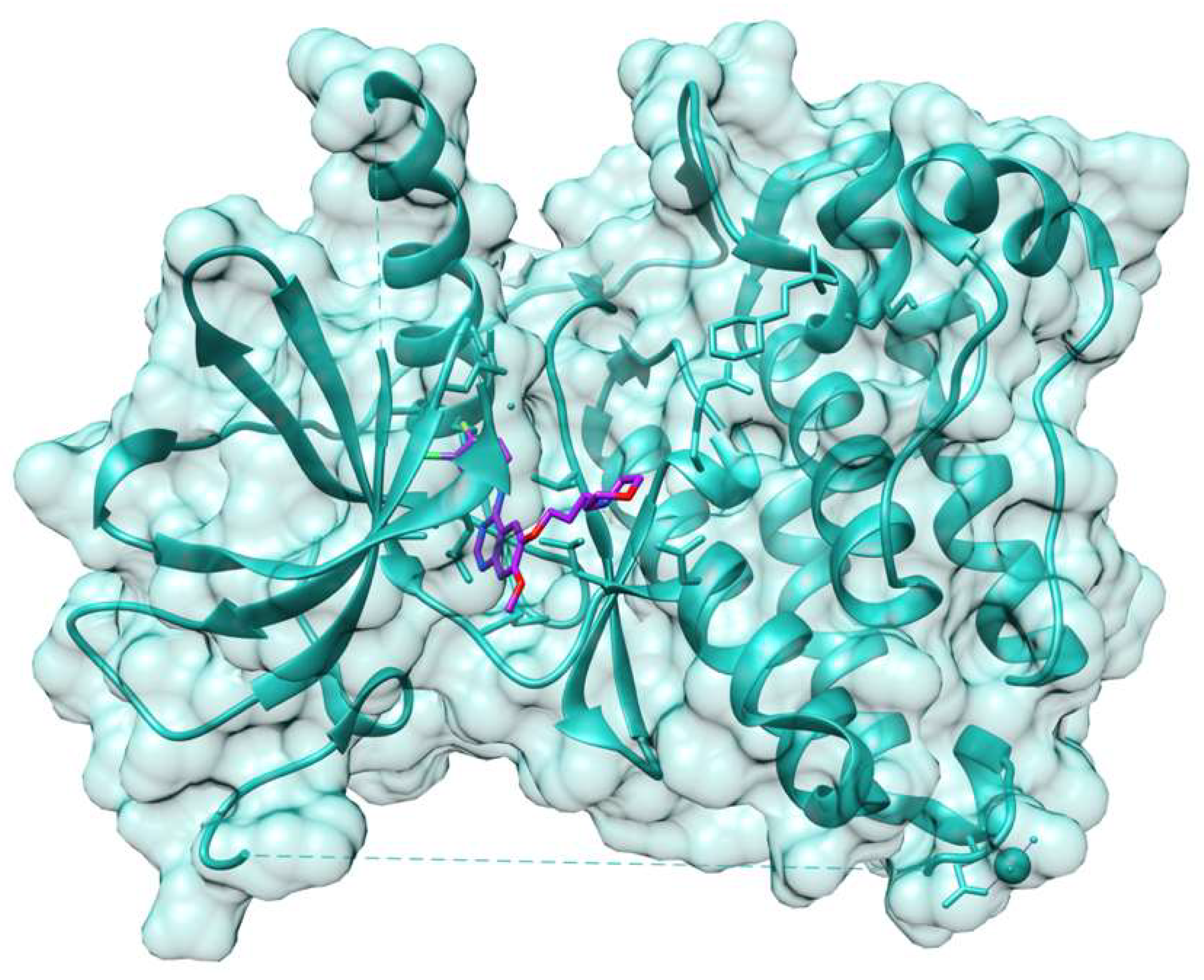
- Mazzorana, M.; Shotton, E.J.; Hall, D.R. A comprehensive approach to X-ray crystallography for drug discovery at a synchrotron facility—The example of Diamond Light Source. Drug Discov. Today Technol. 2020, in press. [Google Scholar] [CrossRef]
- Stiers, K.M.; Graham, A.C.; Zhu, J.S.; Jakeman, D.L.; Nix, J.C.; Beamer, L.J. Structural and dynamical description of the enzymatic reaction of a phosphohexomutase. Struct. Dyn. 2019, 6, 024703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortoli, M.; Torsello, M.; Bickelhaupt, F.M.; Orian, L. Role of the Chalcogen (S, Se, Te) in the Oxidation Mechanism of the Glutathione Peroxidase Active Site. ChemPhysChem 2017, 18, 2990–2998. [Google Scholar] [CrossRef] [PubMed]
- Gołowicz, D.; Kasprzak, P.; Orekhov, V.; Kazimierczuk, K. Fast time-resolved NMR with non-uniform sampling. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 116, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Horitani, M.; Kusubayashi, K.; Oshima, K.; Yato, A.; Sugimoto, H.; Watanabe, K. X-ray Crystallography and Electron Paramagnetic Resonance Spectroscopy Reveal Active Site Rearrangement of Cold-Adapted Inorganic Pyrophosphatase. Sci. Rep. 2020, 10, 4368. [Google Scholar] [CrossRef] [Green Version]
- Zonta, F.; Pagano, M.A.; Trentin, L.; Tibaldi, E.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Pavan, V.; Ribaudo, G.; et al. Lyn sustains oncogenic signaling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood 2015, 125, 3747–3755. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, G. CHAPTER 1: Protein-Protein Interaction Interfaces and their Functional Implications. RSC Drug Discov. Ser. 2021, 1–24. [Google Scholar] [CrossRef]
- Paul, D.; Sanap, G.; Shenoy, S.; Kalyane, D.; Kalia, K.; Tekade, R.K. Artificial intelligence in drug discovery and development. Drug Discov. Today 2021, 26, 80–93. [Google Scholar] [CrossRef]
- Chen, H.; Engkvist, O.; Wang, Y.; Olivecrona, M.; Blaschke, T. The rise of deep learning in drug discovery. Drug Discov. Today 2018, 23, 1241–1250. [Google Scholar] [CrossRef]
- Klambauer, G.; Hochreiter, S.; Rarey, M. Machine Learning in Drug Discovery. J. Chem. Inf. Model. 2019, 59, 945–946. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Klambauer, G.; Unterthiner, T.; Hochreiter, S. DeepTox: Toxicity Prediction using Deep Learning. Front. Environ. Sci. 2016, 3, 80. [Google Scholar] [CrossRef] [Green Version]
- Wainberg, M.; Merico, D.; Delong, A.; Frey, B.J. Deep learning in biomedicine. Nat. Biotechnol. 2018, 36, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tan, J.; Han, D.; Zhu, H. From machine learning to deep learning: Progress in machine intelligence for rational drug discovery. Drug Discov. Today 2017, 22, 1680–1685. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in de novo drug design: From conventional to machine learning methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef]
- Segler, M.H.S.; Waller, M.P. Neural-Symbolic Machine Learning for Retrosynthesis and Reaction Prediction. Chem.-Eur. J. 2017, 23, 5966–5971. [Google Scholar] [CrossRef]
- Coley, C.W.; Barzilay, R.; Jaakkola, T.S.; Green, W.H.; Jensen, K.F. Prediction of Organic Reaction Outcomes Using Machine Learning. ACS Cent. Sci. 2017, 3, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Kalra, G.; Wilson, W.; Peng, Y.; Augsburger, L.L. A Prototype Intelligent Hybrid System for Hard Gelatin Capsule Formulation Development. Pharm. Technol. 2002, 26, 44–60. [Google Scholar] [CrossRef]
- Mehta, C.H.; Narayan, R.; Nayak, U.Y. Computational modeling for formulation design. Drug Discov. Today 2019, 24, 781–788. [Google Scholar] [CrossRef]
- Zhao, C.; Jain, A.; Hailemariam, L.; Suresh, P.; Akkisetty, P.; Joglekar, G.; Venkatasubramanian, V.; Reklaitis, G.V.; Morris, K.; Basu, P. Toward intelligent decision support for pharmaceutical product development. J. Pharm. Innov. 2006, 1, 23–35. [Google Scholar] [CrossRef]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Cheminform. 2017, 9, 42. [Google Scholar] [CrossRef]
- Lenselink, E.B.; ten Dijke, N.; Bongers, B.; Papadatos, G.; van Vlijmen, H.W.T.; Kowalczyk, W.; IJzerman, A.P.; van Westen, G.J.P. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform. 2017, 9, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernández-Lobato, J.M.; Sánchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Kadurin, A.; Nikolenko, S.; Khrabrov, K.; Aliper, A.; Zhavoronkov, A. druGAN: An Advanced Generative Adversarial Autoencoder Model for de Novo Generation of New Molecules with Desired Molecular Properties in Silico. Mol. Pharm. 2017, 14, 3098–3104. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, T.; Olivecrona, M.; Engkvist, O.; Bajorath, J.; Chen, H. Application of Generative Autoencoder in De Novo Molecular Design. Mol. Inform. 2018, 37, 1700123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Mariya, P.; Olexandr, I.; Alexander, T. Deep reinforcement learning for de novo drug design. Sci. Adv. 2021, 4, eaap7885. [Google Scholar] [CrossRef] [Green Version]
- Schütt, K.T.; Gastegger, M.; Tkatchenko, A.; Müller, K.-R.; Maurer, R.J. Unifying machine learning and quantum chemistry with a deep neural network for molecular wavefunctions. Nat. Commun. 2019, 10, 5024. [Google Scholar] [CrossRef] [Green Version]
- Bennett, W.F.D.; He, S.; Bilodeau, C.L.; Jones, D.; Sun, D.; Kim, H.; Allen, J.E.; Lightstone, F.C.; Ingólfsson, H.I. Predicting Small Molecule Transfer Free Energies by Combining Molecular Dynamics Simulations and Deep Learning. J. Chem. Inf. Model. 2020, 60, 5375–5381. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Mirdita, M.; Ovchinnikov, S.; Steinegger, M. ColabFold-Making protein folding accessible to all. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.-M.; Zietz, M.; Hoffman, M.M.; et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 2018, 15, 20170387. [Google Scholar] [CrossRef] [Green Version]
- Matar, P.; Rojo, F.; Cassia, R.; Moreno-Bueno, G.; Di Cosimo, S.; Tabernero, J.; Guzmán, M.; Rodriguez, S.; Arribas, J.; Palacios, J.; et al. Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor Gefitinib (ZD1839) and the monoclonal antibody Cetuximab (IMC-C225): Superiority over single-agent receptor targeting. Clin. Cancer Res. 2004, 10, 6487–6501. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W. Therapeutic development and current uses of BCL-2 inhibition. Hematol. Am. Soc. Hematol. Educ. Program Book 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pagano, M.A.; Tibaldi, E.; Molino, P.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Ribaudo, G.; Leanza, L.; Peruzzo, R.; et al. Mitochondrial apoptosis is induced by Alkoxy phenyl-1-propanone derivatives through PP2A-mediated dephosphorylation of Bad and Foxo3A in CLL. Leukemia 2019, 33, 1148–1160. [Google Scholar] [CrossRef]
- Hughes, T.B.; Miller, G.P.; Swamidass, S.J. Modeling Epoxidation of Drug-like Molecules with a Deep Machine Learning Network. ACS Cent. Sci. 2015, 1, 168–180. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef]
- Beger, R.D.; Schmidt, M.A.; Kaddurah-Daouk, R. Current concepts in pharmacometabolomics, biomarker discovery, and precision medicine. Metabolites 2020, 10, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccia, S.; Liu, J.; Demirkan, A.; van Duijn, C.; Mariani, M.; Castagna, C.; Pastorino, R.; Fiatal, S.; Pikó, P.; Ádány, R.; et al. Identification of Biomarkers for the Prevention of Chronic Disease. SpringerBriefs Public Health 2021, 9–32. [Google Scholar] [CrossRef]
- Upadhyayula, P.S.; Spinazzi, E.F.; Argenziano, M.G.; Canoll, P.; Bruce, J.N. Convection Enhanced Delivery of Topotecan for Gliomas: A Single-Center Experience. Pharmaceutics 2021, 13, 39. [Google Scholar] [CrossRef]
- Wu, S.-K.; Tsai, C.-L.; Huang, Y.; Hynynen, K. Focused Ultrasound and Microbubbles-Mediated Drug Delivery to Brain Tumor. Pharmaceutics 2021, 13, 15. [Google Scholar] [CrossRef]
- Griffith, J.I.; Rathi, S.; Zhang, W.; Zhang, W.; Drewes, L.R.; Sarkaria, J.N.; Elmquist, W.F. Addressing BBB Heterogeneity: A New Paradigm for Drug Delivery to Brain Tumors. Pharmaceutics 2020, 12, 1205. [Google Scholar] [CrossRef] [PubMed]
- Zonta, N.; Cozza, G.; Gianoncelli, A.; Korb, O.; Exner, T.E.; Meggio, F.; Zagotto, G.; Moro, S. Scouting novel protein kinase A (PKA) inhibitors by using a consensus docking-based virtual screening approach. Lett. Drug Des. Discov. 2009, 6, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Straehla, J.P.; Warren, K.E. Pharmacokinetic Principles and Their Application to Central Nervous System Tumors. Pharmaceutics 2020, 12, 948. [Google Scholar] [CrossRef]
- Tosi, U.; Souweidane, M. Convection Enhanced Delivery for Diffuse Intrinsic Pontine Glioma: Review of a Single Institution Experience. Pharmaceutics 2020, 12, 660. [Google Scholar] [CrossRef] [PubMed]
- Molotkov, A.; Carberry, P.; Dolan, M.A.; Joseph, S.; Idumonyi, S.; Oya, S.; Castrillon, J.; Konofagou, E.E.; Doubrovin, M.; Lesser, G.J.; et al. Real-Time Positron Emission Tomography Evaluation of Topotecan Brain Kinetics after Ultrasound-Mediated Blood–Brain Barrier Permeability. Pharmaceutics 2021, 13, 405. [Google Scholar] [CrossRef]
- Chatterjee, K.; Atay, N.; Abler, D.; Bhargava, S.; Sahoo, P.; Rockne, R.C.; Munson, J.M. Utilizing Dynamic Contrast-Enhanced Magnetic Resonance Imaging (DCE-MRI) to Analyze Interstitial Fluid Flow and Transport in Glioblastoma and the Surrounding Parenchyma in Human Patients. Pharmaceutics 2021, 13, 212. [Google Scholar] [CrossRef]
- Sharabi, S.; Last, D.; Daniels, D.; Fabian, I.D.; Atrakchi, D.; Bresler, Y.; Liraz-Zaltsman, S.; Cooper, I.; Mardor, Y. Non-Invasive Low Pulsed Electrical Fields for Inducing BBB Disruption in Mice—Feasibility Demonstration. Pharmaceutics 2021, 13, 169. [Google Scholar] [CrossRef]
- Nwagwu, C.D.; Immidisetti, A.V.; Bukanowska, G.; Vogelbaum, M.A.; Carbonell, A.-M. Convection-Enhanced Delivery of a First-in-Class Anti-β1 Integrin Antibody for the Treatment of High-Grade Glioma Utilizing Real-Time Imaging. Pharmaceutics 2021, 13, 40. [Google Scholar] [CrossRef]
- Brady, M.; Raghavan, R.; Sampson, J. Determinants of Intraparenchymal Infusion Distributions: Modeling and Analyses of Human Glioblastoma Trials. Pharmaceutics 2020, 12, 895. [Google Scholar] [CrossRef] [PubMed]
- Binaschi, M.; Zagotto, G.; Palumbo, M.; Zunino, F.; Farinosi, R.; Capranico, G. Irreversible and reversible topoisomerase II DNA cleavage stimulated by clerocidin: Sequence specificity and structural drug determinants. Cancer Res. 1997, 57, 1710–1716. [Google Scholar] [PubMed]
- Mehta, J.N.; McRoberts, G.R.; Rylander, C.G. Controlled Catheter Movement Affects Dye Dispersal Volume in Agarose Gel Brain Phantoms. Pharmaceutics 2020, 12, 753. [Google Scholar] [CrossRef] [PubMed]
- Bienfait, K.; Chhibber, A.; Marshall, J.C.; Armstrong, M.; Cox, C.; Shaw, P.M.; Paulding, C. Current challenges and opportunities for pharmacogenomics: Perspective of the Industry Pharmacogenomics Working Group (I-PWG). Hum. Genet. 2021. [Google Scholar] [CrossRef] [PubMed]
- Peiró, A.M. Pharmacogenetics in Pain Treatment. Adv. Pharmacol. 2018, 83, 247–273. [Google Scholar] [CrossRef]
- DeWeerdt, S. Tracing the US opioid crisis to its roots. Nature 2019, 573, S10–S12. [Google Scholar] [CrossRef] [Green Version]
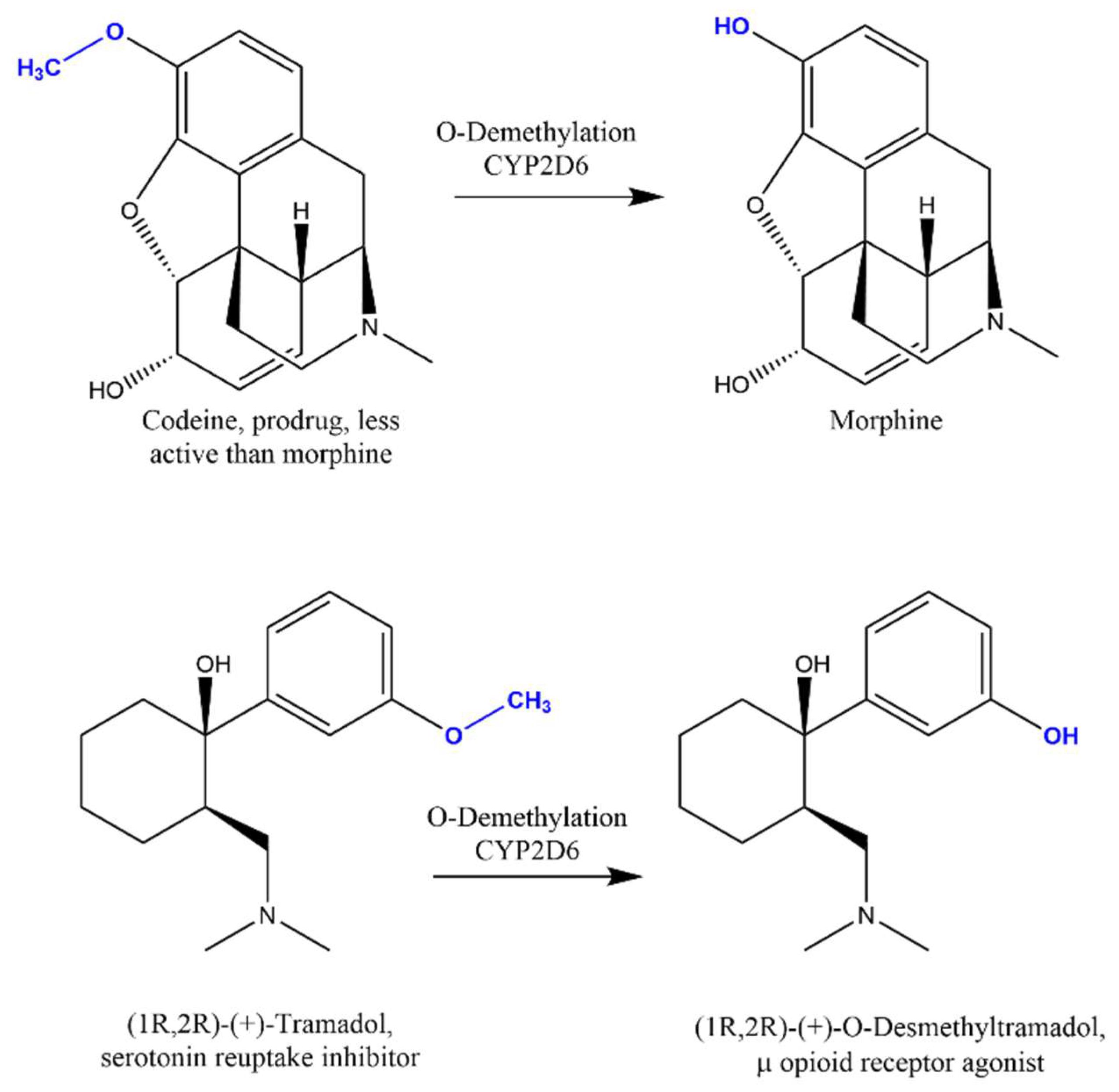
- Gammal, R.S.; Crews, K.R.; Haidar, C.E.; Hoffman, J.M.; Baker, D.K.; Barker, P.J.; Estepp, J.H.; Pei, D.; Broeckel, U.; Wang, W.; et al. Pharmacogenetics for Safe Codeine Use in Sickle Cell Disease. Pediatrics 2016, 138, e20153479. [Google Scholar] [CrossRef] [Green Version]
- Benjeddou, M.; Peiró, A.M. Pharmacogenomics and prescription opioid use. Pharmacogenomics 2021, 22, 235–245. [Google Scholar] [CrossRef]
- Tremaine, L.; Delmonte, T.; Francke, S. The role of ADME pharmacogenomics in early clinical trials: Perspective of the of the Industry Pharmacogenomics Working Group (I-PWG). Pharmacogenomics 2015, 16, 2055–2067. [Google Scholar] [CrossRef] [Green Version]
- Relling, M.V.; Klein, T.E.; Gammal, R.S.; Whirl-Carrillo, M.; Hoffman, J.M.; Caudle, K.E. The Clinical Pharmacogenetics Implementation Consortium: 10 Years Later. Clin. Pharmacol. Ther. 2020, 107, 171–175. [Google Scholar] [CrossRef]
- Chenoweth, M.J.; Giacomini, K.M.; Pirmohamed, M.; Hill, S.L.; van Schaik, R.H.N.; Schwab, M.; Shuldiner, A.R.; Relling, M.V.; Tyndale, R.F. Global Pharmacogenomics Within Precision Medicine: Challenges and Opportunities. Clin. Pharmacol. Ther. 2020, 107, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J. The latest automated docking technologies for novel drug discovery. Expert Opin. Drug Discov. 2021, 16, 625–645. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zagotto, G.; Bortoli, M. Drug Design: Where We Are and Future Prospects. Molecules 2021, 26, 7061. https://doi.org/10.3390/molecules26227061
Zagotto G, Bortoli M. Drug Design: Where We Are and Future Prospects. Molecules. 2021; 26(22):7061. https://doi.org/10.3390/molecules26227061
Chicago/Turabian StyleZagotto, Giuseppe, and Marco Bortoli. 2021. "Drug Design: Where We Are and Future Prospects" Molecules 26, no. 22: 7061. https://doi.org/10.3390/molecules26227061
APA StyleZagotto, G., & Bortoli, M. (2021). Drug Design: Where We Are and Future Prospects. Molecules, 26(22), 7061. https://doi.org/10.3390/molecules26227061