
Elucidating the Glucokinase Activating Potentials of Naturally Occurring Prenylated Flavonoids: An Explicit Computational Approach

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Results and Discussion
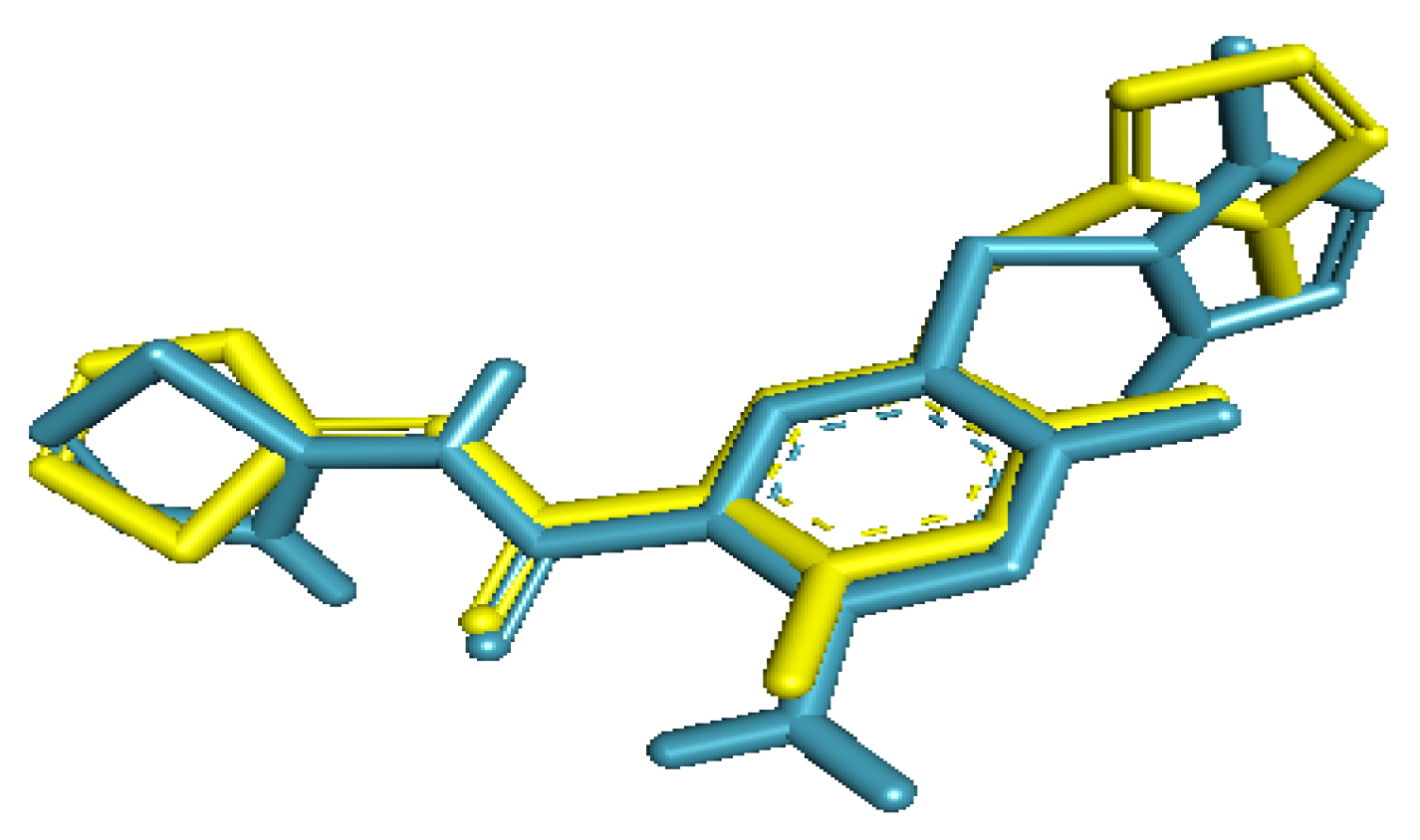
2.1. Validation of Docking Protocol
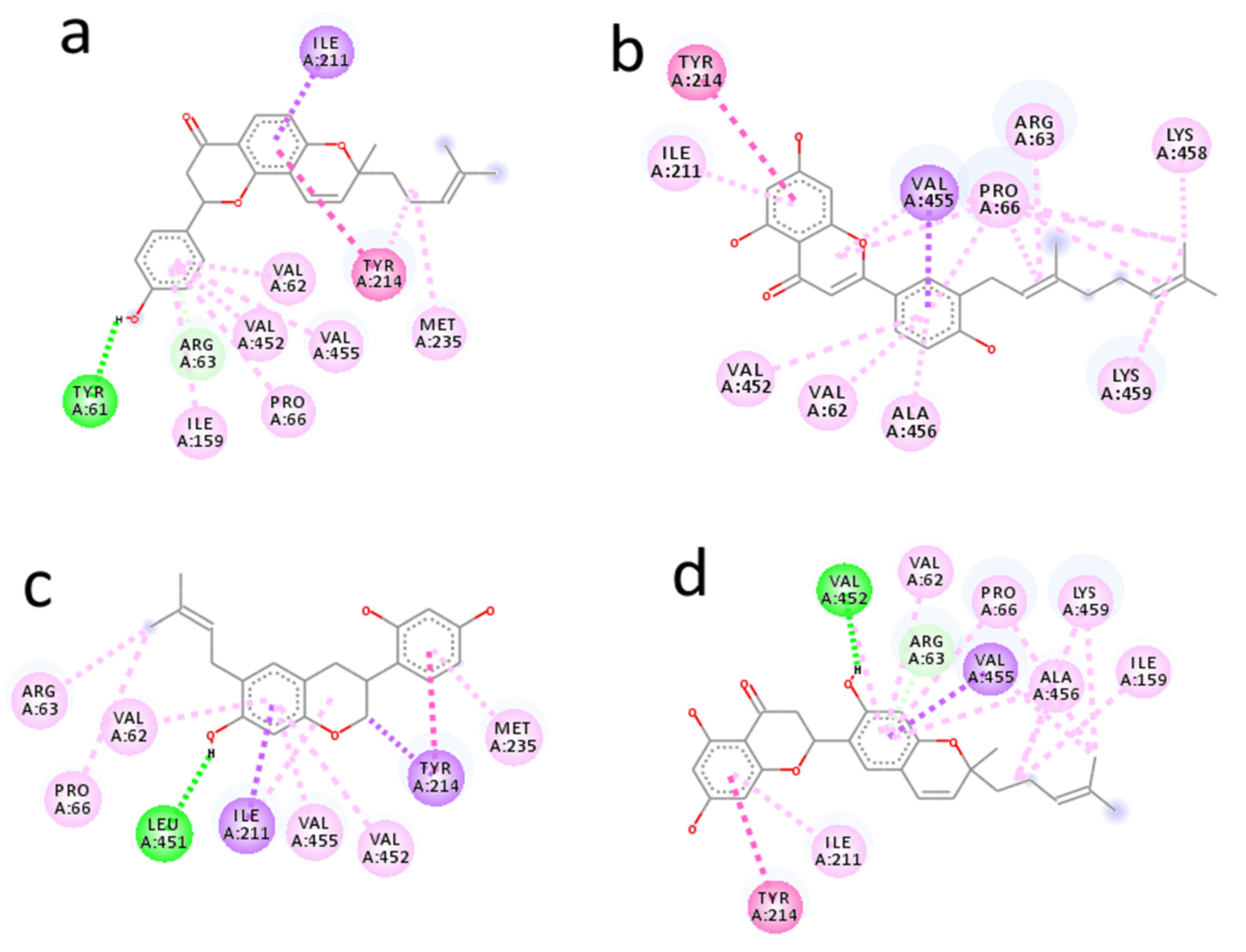
2.2. Molecular Docking Analysis of Prenylated Flavonoids with Glucokinase Enzyme
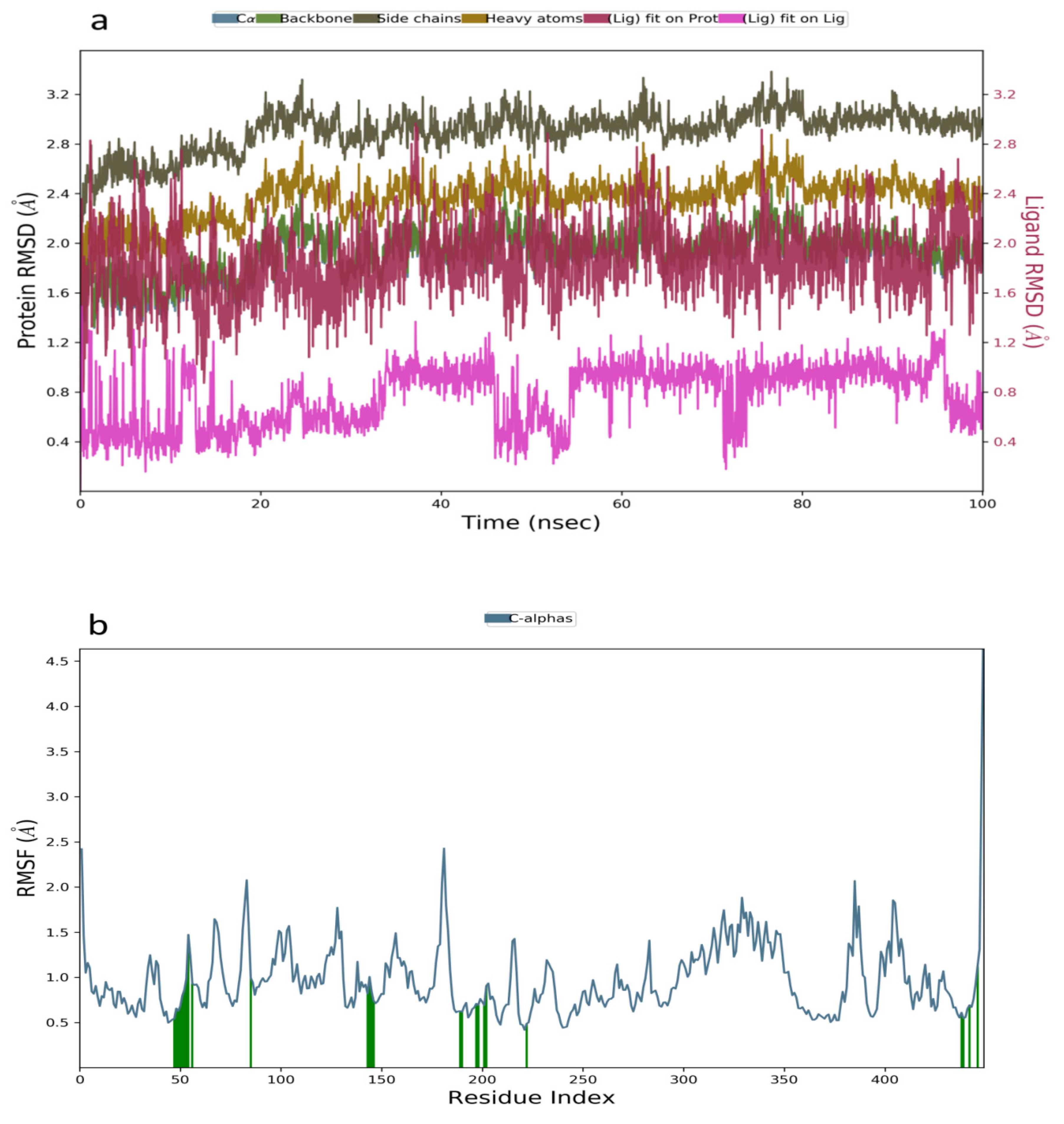
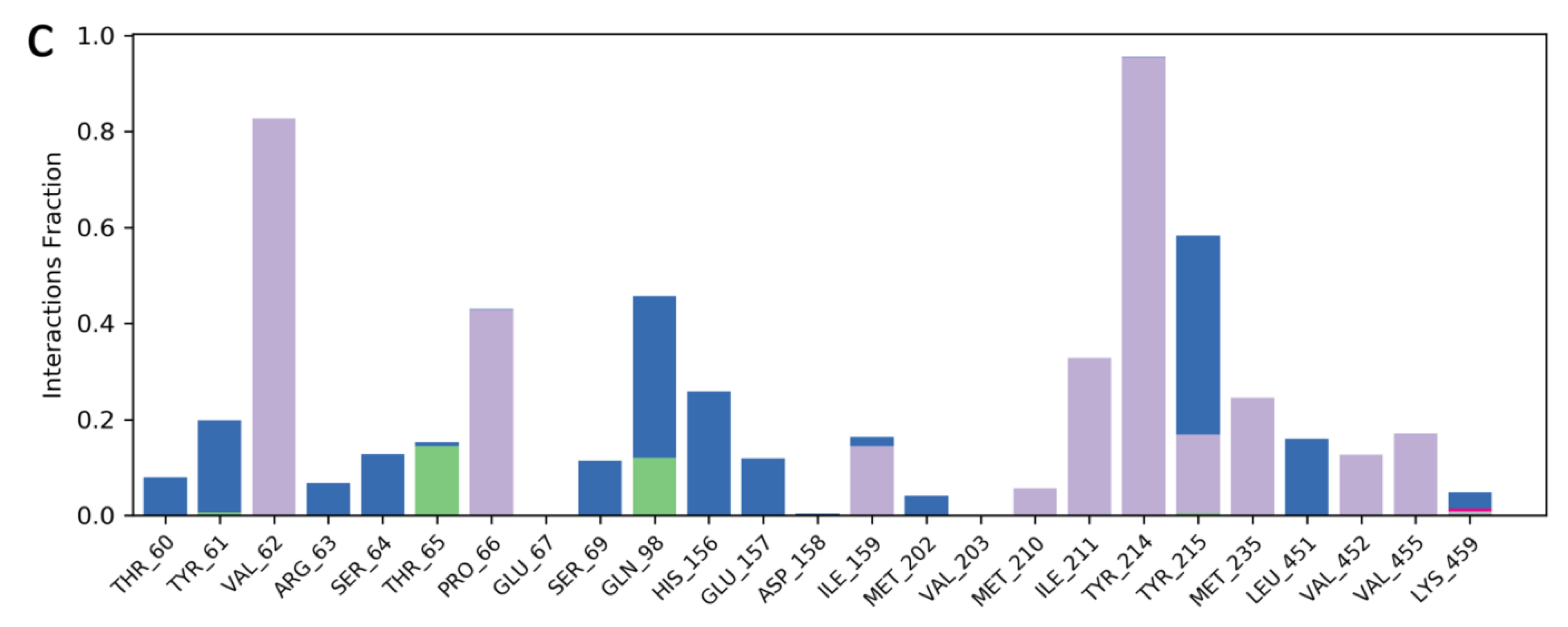
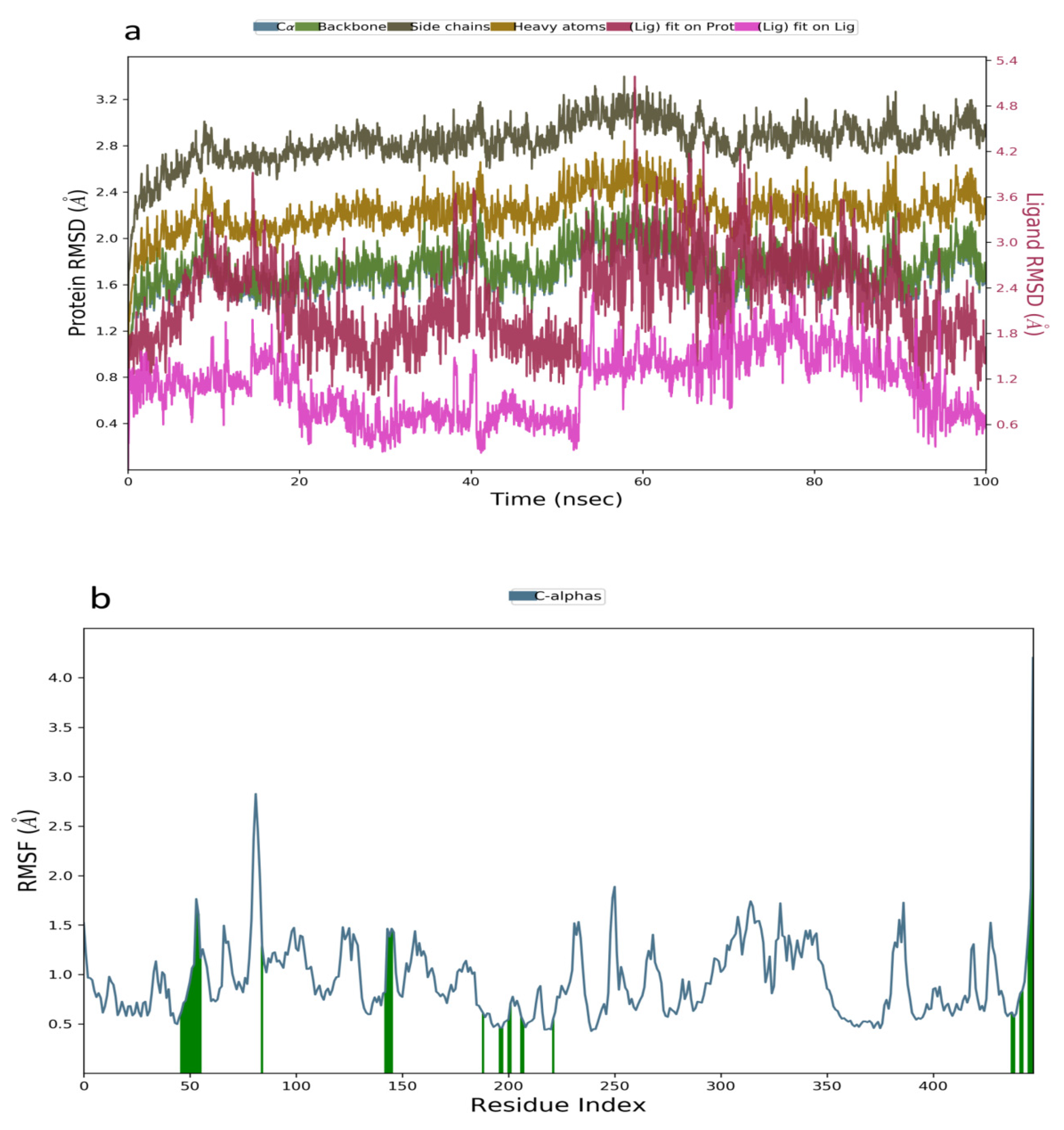
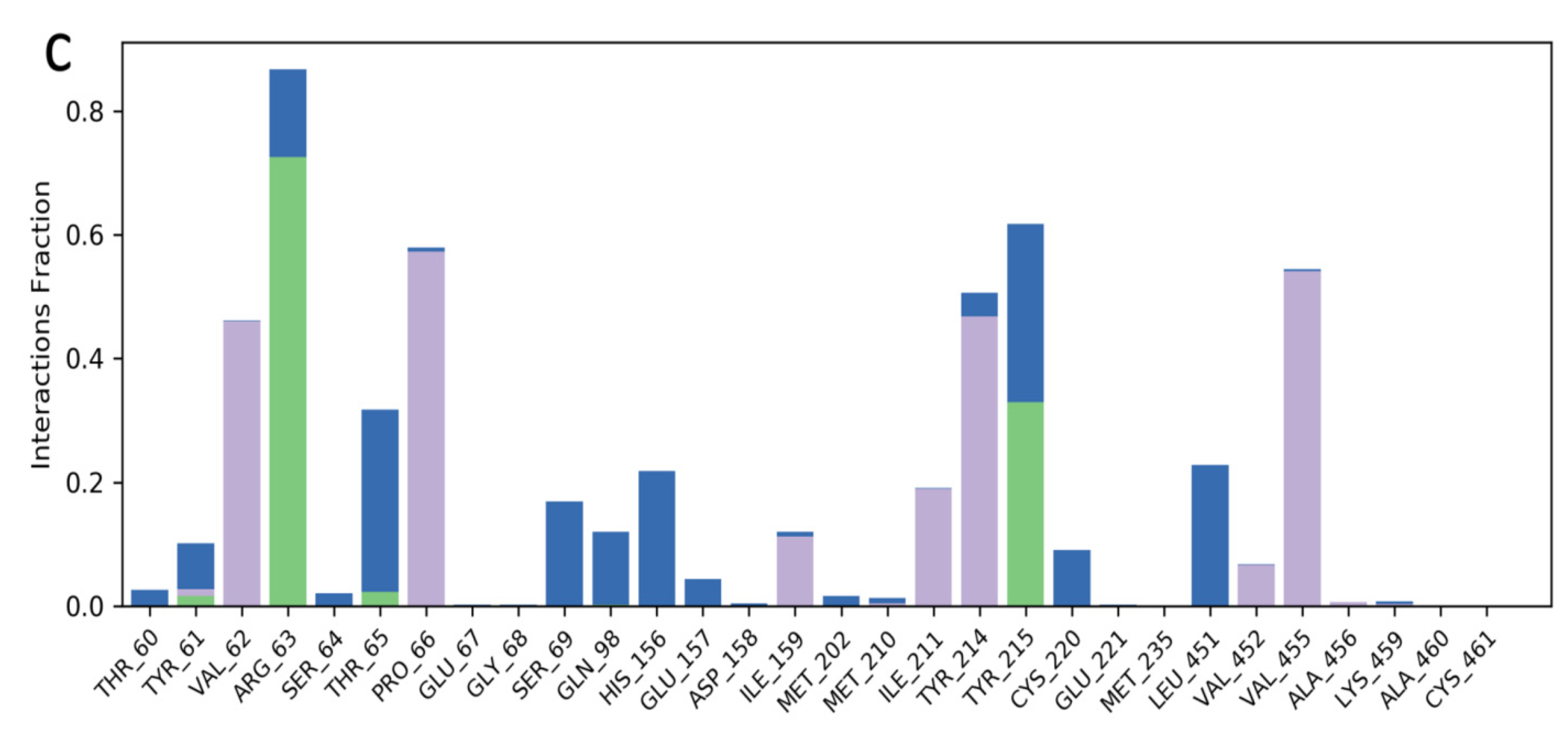
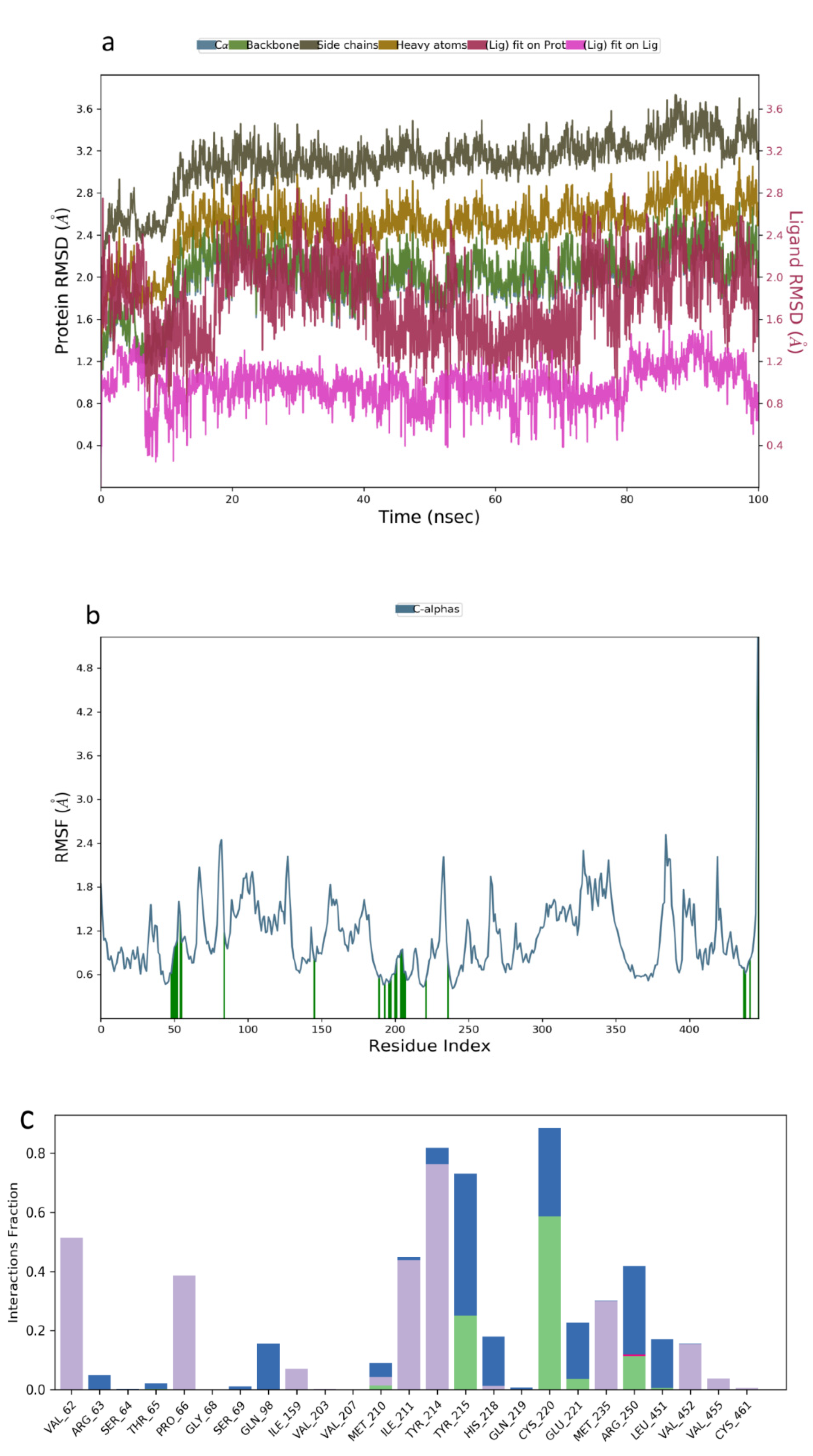
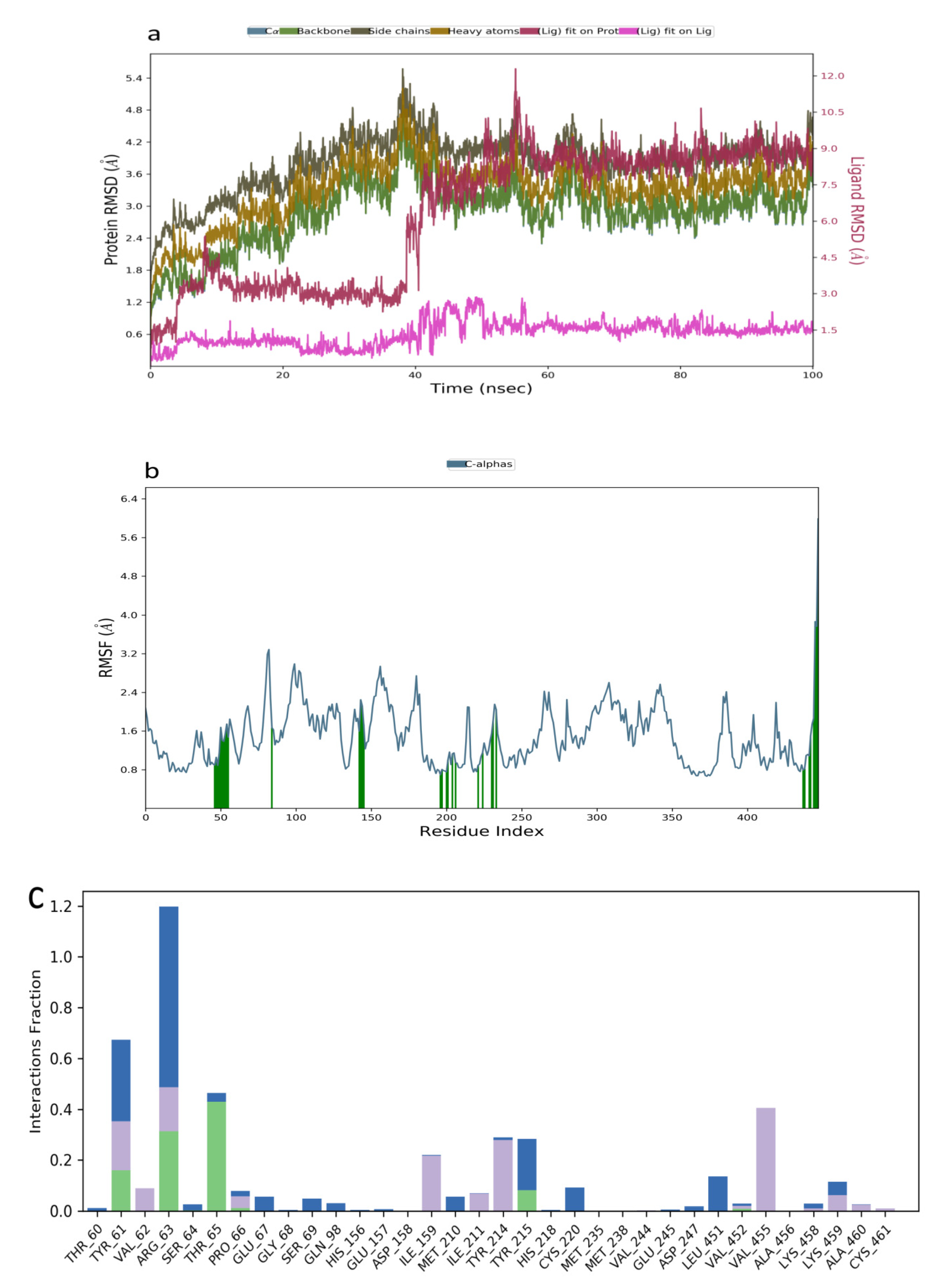
2.3. Molecular Dynamics Simulation
2.4. Analysis of Binding Free Energy
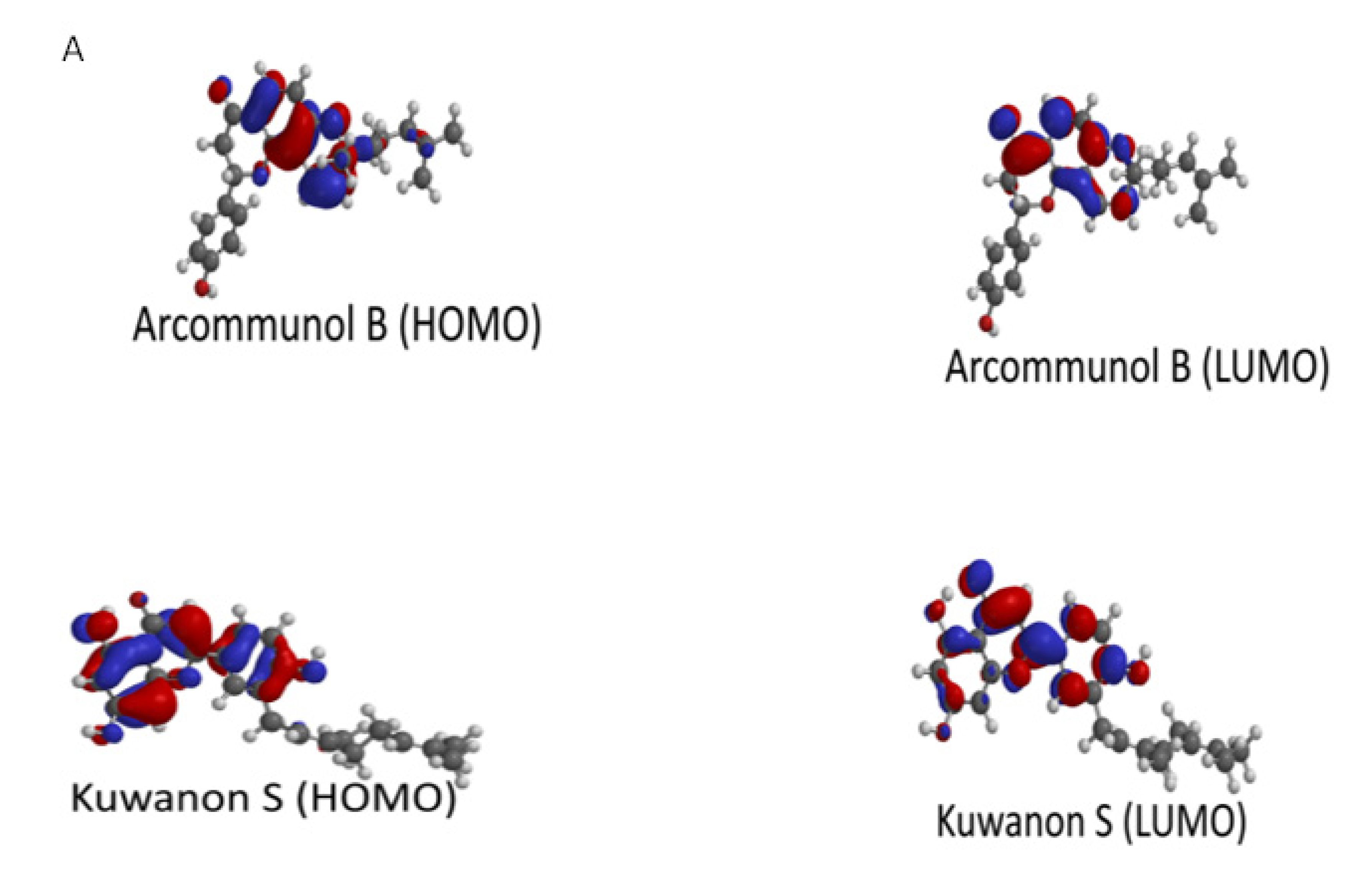
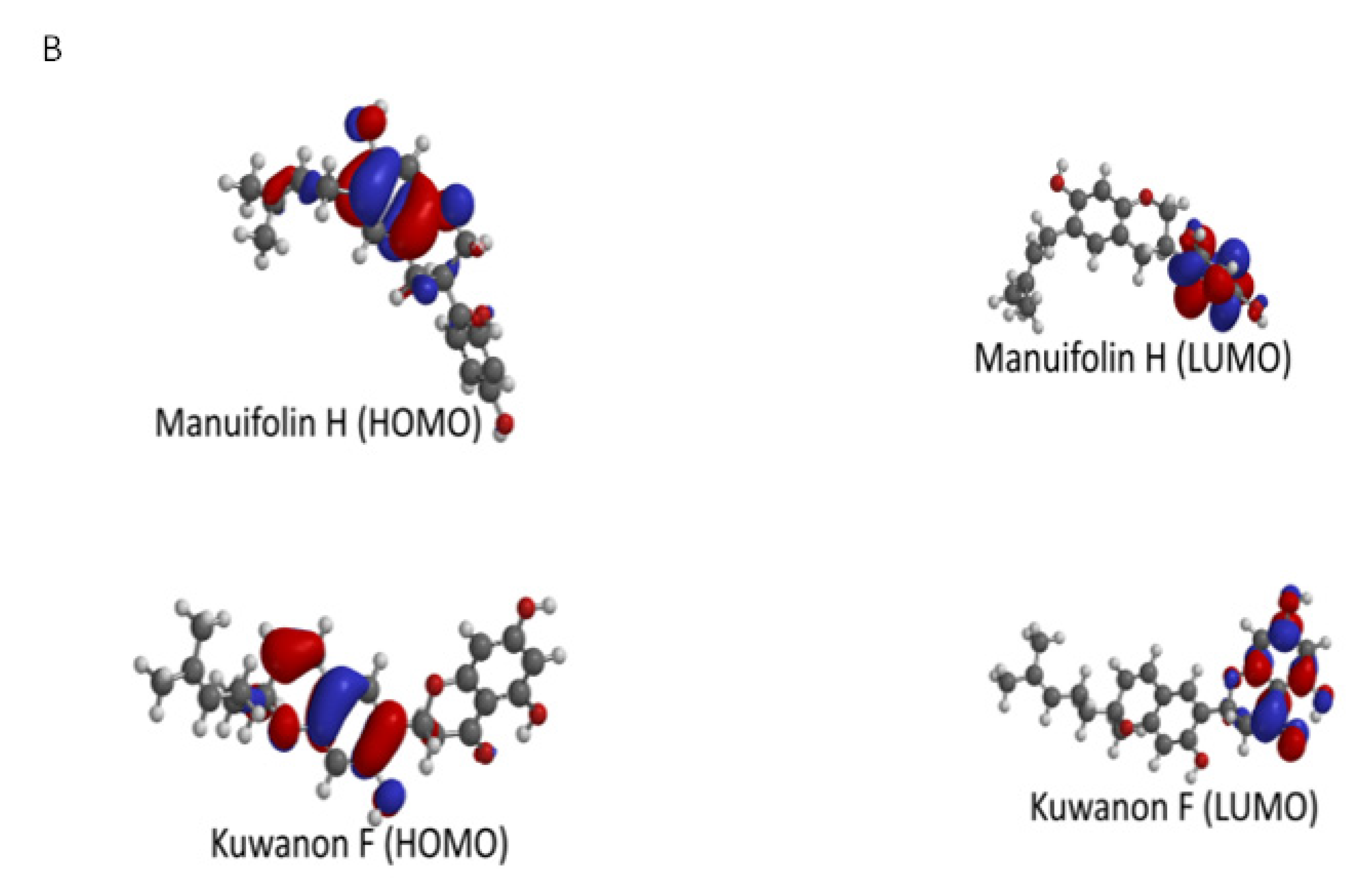
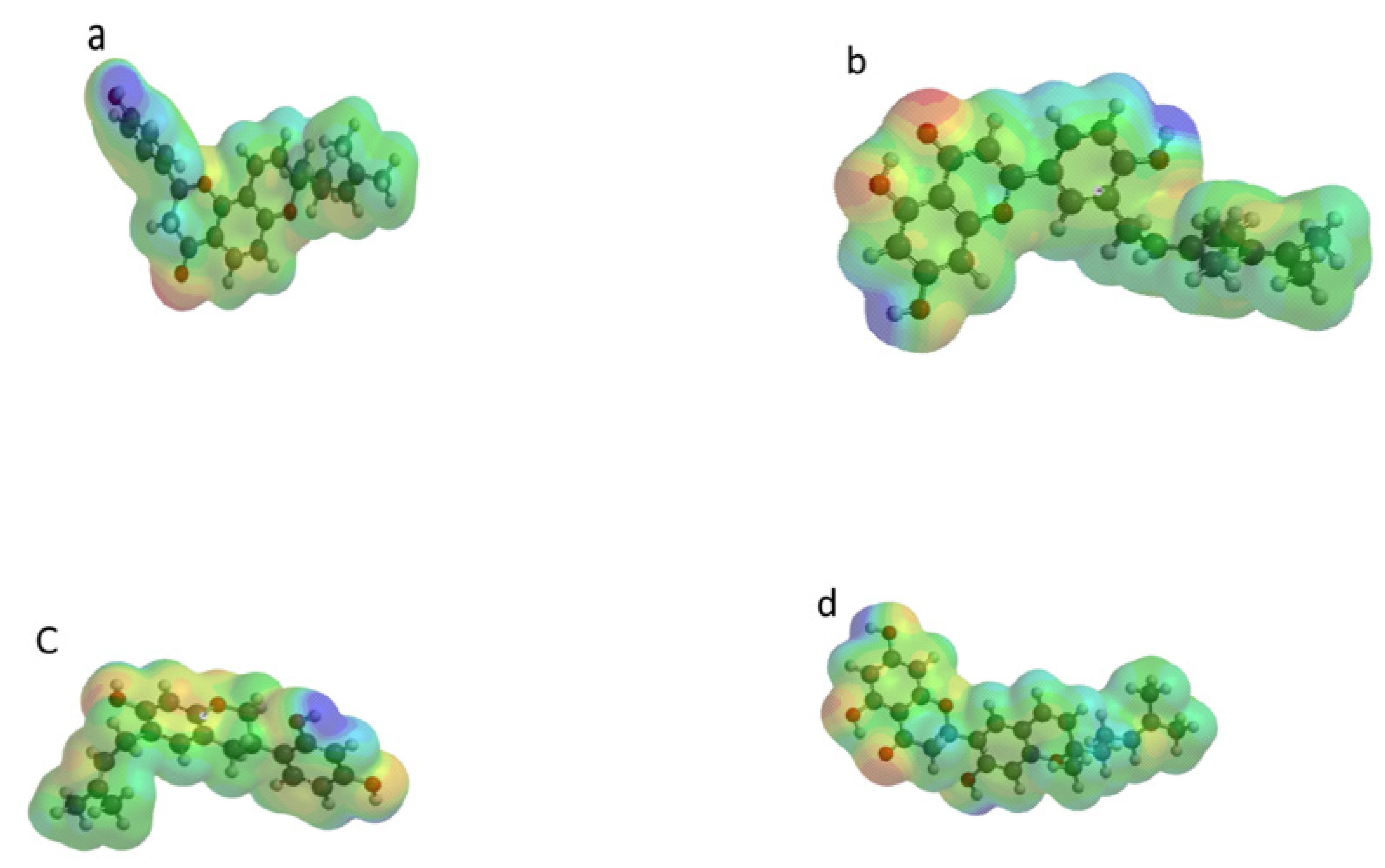
2.5. Molecular Modeling and Quantum Chemical Calculations
2.6. Pharmacokinetic and Drug-Likeness Studies
3. Materials and Methods
3.1. Receptor and Ligand Preparation
3.2. Validation of Docking Protocol and Molecular Docking Studies
3.3. Molecular Dynamics Simulation
3.4. Theoretical Modelling and Optimization Studies
3.5. Pharmacokinetic and Drug-Likeness Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Matschinsky, F.M.; Wilson, D.F. The central role of glucokinase in glucose homeostasis: A perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front. Physiol. 2019, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Remedi, M.S.; Koster, J.C.; Patton, B.L.; Nichols, C.G. ATP-sensitive K+ channel signaling in glucokinase-deficient diabetes. Diabetes 2005, 54, 2925–2931. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Dunn-Meynell, A.A.; Routh, V.H.; Gaspers, L.D.; Nagata, Y.; Nishimura, T.; Eiki, J.; Zhang, J.; Levin, B.E. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes 2006, 55, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, R.; Kubota, H.; Yugi, K.; Toyoshima, Y.; Komori, Y.; Soga, T.; Kuroda, S. The selective control of glycolysis, gluconeogenesis and glycogenesis by temporal insulin patterns. Mol. Syst. Biol. 2013, 9, 664. [Google Scholar] [CrossRef]
- Efeyan, A.; Comb, W.C.; Sabatini, D.M. Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Njølstad, P.R.; Søvik, O.; Cuesta-Muñoz, A.; Bjørkhaug, L.; Massa, O.; Barbetti, F.; Undlien, D.E.; Shiota, C.; Magnuson, M.A.; Molven, A.; et al. Neonatal diabetes mellitus due to complete glucokinase deficiency. N. Engl. J. Med. 2001, 344, 1588–1592. [Google Scholar] [CrossRef]
- Agius, L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem. J. 2008, 414, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Matschinsky, F.M.; Zelent, B.; Doliba, N.M.; Kaestner, K.H.; Vanderkooi, J.M.; Grimsby, J.; Sarabu, R. Research and development of glucokinase activators for diabetes therapy: Theoretical and practical aspects. Diabetes-Perspect. Drug Ther. 2011, 203, 357–401. [Google Scholar]
- Yellapu, N.K.; Kandlapalli, K.; Kandimalla, R.; Adi, P.J. Conformational transition pathway of R308K mutant glucokinase in the presence of the glucokinase activator YNKGKA 4. FEBS Open Biol. 2018, 8, 1202–1208. [Google Scholar] [CrossRef]
- Toulis, K.A.; Nirantharakumar, K.; Pourzitaki, C.; Barnett, A.H.; Tahrani, A.A. Glucokinase activators for type 2 diabetes: Challenges and future developments. Drugs 2020, 80, 467–475. [Google Scholar] [CrossRef]
- Amin, N.B.; Aggarwal, N.; Pall, D.; Paragh, G.; Denney, W.S.; Le, V.; Riggs, M.; Calle, R.A. Two dose-ranging studies with PF−04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Simons, P.I.; Simons, N.; Stehouwer, C.D.; Schalkwijk, C.G.; Schaper, N.C.; Brouwers, M.C. Association of common gene variants in glucokinase regulatory protein with cardiorenal disease: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0206174. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Jiang, Y.; Yang, J.; He, J.; Sun, J.; Chen, F.; Yang, B. Prenylated flavonoids, promising nutraceuticals with impressive biological activities. Trends Food. Sci. Technol. 2015, 44, 93–104. [Google Scholar] [CrossRef]
- Yazaki, K.; Sasaki, K.; Tsurumaru, Y. Prenylation of aromatic compounds, a key diversification of plant secondary metabolites. Phytochemistry 2009, 70, 1739–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, G.; Huhman, D.; Lei, Z.; Snyder, J.; Sumner, L.W.; Dixon, R.A. Characterization of an isoflavonoid-specific prenyltransferase from Lupinus albus. Plant Physiol. 2012, 159, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.J.; Wu, B.; Ding, T.; Chu, J.H.; Li, C.Y.; Zhang, J.; Wu, T.; Wu, J.; Liu, S.J.; Liu, S.L.; et al. Simultaneous characterization of prenylated flavonoids and isoflavonoids in Psoralea corylifolia L. by liquid chromatography with diode array detection and quadrupole time of flight mass spectrometry. RCM 2012, 26, 2343–2358. [Google Scholar] [CrossRef]
- Jhong, C.H.; Riyaphan, J.; Lin, S.H.; Chia, Y.C.; Weng, C.F. Screening alpha-glucosidase and alpha-amylase inhibitors from natural compounds by molecular docking in silico. Biofactors 2015, 41, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Bourjot, M.; Apel, C.; Martin, M.T.; Grellier, P.; Guéritte, F.; Litaudon, M. Antiplasmodial, antitrypanosomal, and cytotoxic activities of prenylated flavonoids isolated from the stem bark of artocarpus styracifolius. Planta Med. 2010, 76, 1600–1604. [Google Scholar] [CrossRef]
- Baldi, A. Computational approaches for drug design and discovery: An overview. Syst. Rev. Pharm. 2010, 1, 99. [Google Scholar] [CrossRef]
- Palermo, G.; De Vivo, M. Computational chemistry for drug discovery. Encycl. Nanotechnol. 2014, 66, 334–395. [Google Scholar]
- Sethi, A.; Joshi, K.; Sasikala, K.; Alvala, M. Molecular docking in modern drug discovery: Principles and recent applications. Drug Discov. Develop. New Adv. 2019, 27–39. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Marinho, M.M.; Almeida-Neto, F.W.Q.; Marinho, E.M.; da Silva, L.P.; Menezes, R.R.; Dos Santos, R.P.; Marinho, E.S.; de Lima-Neto, P.; Martins, A.M. Quantum computational investigations and molecular docking studies on amentoflavone. Heliyon 2021, 7, e06079. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, H.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Mousavi, S.S.; Karami, A.; Haghighi, T.M.; Tumilaar, S.G.; Idroes, R.; Mahmud, S.; Celik, I.; Ağagündüz, D.; Tallei, T.E.; Emran, T.B.; et al. In Silico Evaluation of Iranian Medicinal Plant Phytoconstituents as Inhibitors against Main Protease and the Receptor-Binding Domain of SARS-CoV−2. Molecules 2021, 26, 5724. [Google Scholar] [CrossRef]
- Olasupo, S.B.; Uzairu, A.; Shallangwa, G.; Uba, S. QSAR modeling, molecular docking and ADMET/pharmacokinetic studies: Achemometrics approach to search for novel inhibitors of norepinephrine transporter as potent antipsychotic drugs. J. Iran. Chem. Soc. 2020, 17, 1953–1966. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, V.K.; Singh, R.; Sharma, J.; Das, P.; Purohit, R. Structural based study to identify new potential inhibitors for dualspecificity tyrosine-phosphorylation—Regulated kinase. Comput. Methods Prog. Biomed. 2020, 194, 105494. [Google Scholar] [CrossRef] [PubMed]
- Adewole, S.O.; Ojewole, J.A. Artocarpus communis Forst. root-bark aqueous extract and streptozotocin-induced ultrastructural and metabolic changes in hepatic tissues of Wistar rats. Afr. J. Tradit. 2007, 4, 397. [Google Scholar]
- Lans, C.A. Ethnomedicines used in Trinidad and Tobago for urinary problems and diabetes mellitus. J. Ethnobiol Ethnomed. 2006, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Angadi, K.K.; Gundampati, R.K.; Jagannadham, M.V.; Kandru, A. Molecular docking studies of guggultetrol from Nymphaea pubescens with target glucokinase (GK) related to type-II Diabetes. J. Appl. Pharm. Sci. 2013, 3, 127. [Google Scholar]
- Damián-Medina, K.; Salinas-Moreno, Y.; Milenkovic, D.; Figueroa-Yáñez, L.; Marino-Marmolejo, E.; Higuera-Ciapara, I.; Lugo-Cervantes, E. In silico analysis of antidiabetic potential of phenolic compounds from blue corn (Zea mays L.) and black bean (Phaseolus vulgaris L.). Heliyon 2020, 6, e03632. [Google Scholar] [CrossRef]
- Grewal, A.S.; Arora, S.; Sharma, N.; Singh, S. In silico docking studies of compounds from Persian shallot as allosteric glucokinase activators. Plant Arch. 2020, 20, 3768–3771. [Google Scholar]
- Uniyal, A.; Mahapatra, M.K.; Tiwari, V.; Sandhir, R.; Kumar, R. Targeting SARS-CoV−2 main protease: Structure based virtual screening, in silico ADMET studies and molecular dynamics simulation for identification of potential inhibitors. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ayeni, A.O.; Akinyele, O.F.; Hosten, E.C.; Fakola, E.G.; Olalere, J.T.; Egharevba, G.O.; Watkins, G.M. Synthesis, crystal structure, experimental and theoretical studies of corrosion inhibition of 2-((4-(2-hydroxy−4-methylbenzyl) piperazin−1-yl) methyl)−5-methylphenol–A Mannich base. J. Mol. Struct. 2020, 1219, 128539. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pandey, A.K.; Jain, S.; Misra, N. FT-IR spectroscopy, intra-molecular C−H⋯ O interactions, HOMO, LUMO, MESP analysis and biological activity of two natural products, triclisine and rufescine: DFT and QTAIM approaches. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 136, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Badry, R.; Ibrahim, A.; Gamal, F.; Shehata, D.; Ezzat, H.; Elhaes, H.; Ibrahim, M. Electronic Properties of Polyvinyl Alcohol/TiO2/SiO2 Nanocomposites. Biointerface Res. Appl. Chem. 2020, 10, 6427–6435. [Google Scholar]
- Tao, Y.; Han, L.; Li, X.; Han, Y.; Liu, Z. Molecular structure, spectroscopy (FT-IR, FT-Raman), thermodynamic parameters, molecular electrostatic potential and HOMO-LUMO analysis of 2,6-dichlorobenzamide. J. Mol. Struct. 2016, 1108, 307–314. [Google Scholar] [CrossRef]
- Bhavani, K.; Renuga, S.; Muthu, S. Quantum mechanical study and spectroscopic (FT-IR, FT-Raman, 13C, 1H) study, first order hyperpolarizability, NBO analysis, HOMO and LUMO analysis of 2-acetoxybenzoic acid by density functional methods. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2015, 136, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, N.; Sundaraganesan, N.; Jayabharathi, J. Molecular structure, spectroscopic (FT-IR, FT-Raman, NMR, UV) studies and first-order molecular hyperpolarizabilities of 1, 2-bis (3-methoxy−4-hydroxybenzylidene) hydrazine by density functional method. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2010, 76, 259–269. [Google Scholar] [CrossRef]
- Obi-Egbedi, N.O.; Essien, K.E.; Obot, I.B.; Ebenso, E.E. 1,2-Diaminoanthraquinone as corrosion inhibitor for mild steel in hydrochloric acid: Weight loss and quantum chemical study. Int. J. Electro.Sci. 2011, 6, 913–930. [Google Scholar]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [Green Version]
- Stefaniu, A.; Pintilie, L. Molecular descriptors and properties of organic molecules. In Symmetry (Group Theory) and Mathematical Treatment in Chemistry; InTech: Rijeka, Croatia, 2018; pp. 161–176. [Google Scholar]
- Ogboye, R.M.; Patil, R.B.; Famuyiwa, S.O.; Faloye, K.O. Novel α-amylase and α-glucosidase inhibitors from selected Nigerian antidiabetic plants: An in silico approach. J. Biomol. Struct. Dyn. 2021, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.J.; Chan, J.; Greenwood, M.N.; Popa-Burke, I.; Remlinger, K.S.; Pickett, S.D.; Green, D.V.; Fillmore, M.C.; Dean, T.W.; Luengo, J.I.; et al. Nuisance compounds, PAINS filters, and dark chemical matter in the GSK HTS collection. SLAS DISCOVERY Adv. Life Sci. RD 2018, 23, 532–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar]
- Release, S. Desmond molecular dynamics system, DE Shaw research, New York, NY, 2017. In Maestro-Desmond Interoperability Tools; Schrödinger: New York, NY, USA, 2017. [Google Scholar]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory. Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Toukmaji, A.Y.; Board, J.A., Jr. Ewald summation techniques in perspective: A survey. Comput. Phys. Comm. 1996, 95, 73–92. [Google Scholar] [CrossRef]
- Zielkiewicz, J. Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J. Chem. Phys 2005, 123, 104501. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of HartreeeFock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Lipinski, C.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand Pubchem ID | Chemical Structure | Binding Energy (kcal/mol) | Hydrogen Bonding Interaction | Hydrophobic Interaction | Pi-Interaction | |
|---|---|---|---|---|---|---|
| Amino Acid Residue | Distance (Å) | |||||
| Arcommunol B 101781179 |  | −10.1 | Tyr61 | 2.19 | Val62, Arg63, Pro66, Ile159, Ile211, Tyr214, Val452, Val455, | Val62, Arg63, Pro66, Ile159, Ile211, Tyr214, Met235, Val452, Val455 |
| Kuwanon S 6450924 |  | −9.6 | - | - | Val62, Arg63, Pro66, Ile211, Tyr214, Val452, Val455, Ala456, Lys458, Lys459, | Val62, Arg63, Pro66, Ile211, Tyr214, Val452, Val455, Ala456, Lys458, Lys459. |
| Manuifolin H 15837463 |  | −9.5 | Leu451 | 2.05 | Val62, Arg63, Pro66, Ile211, Tyr214, Met235, Val452, Val455, | Val62, Arg63, Pro66, Ile211, Tyr214, Met235, Val452, Val455. |
| Kuwanon F 156149 |  | −9.4 | Val452 | 2.82 | Val62, Arg63, Pro66, Ile159, Ile211, Tyr214, Val452, Val455, | Val62, Arg63, Pro66, Ile211, Tyr214, Val452, Val455 |
| Ligand | MMGBSA ΔG Bind | MMGBSA ΔG Bind Coulomb | MMGBSA ΔG Bind Covalent | MMGBSA ΔG Bind Solvation Energy | MMGBSA ΔG Bind vdW |
|---|---|---|---|---|---|
| Arcommunol B | −70.23 | −5.27 | 1.63 | 21.11 | −57.91 |
| Kuwanon S | −54.86 | −9.12 | 2.23 | 17.20 | −50.88 |
| Manuifolin H | −37.34 | −7.26 | 1.35 | 15.94 | −29.89 |
| Kuwanon F | −26.76 | −8.64 | 0.97 | 14.48 | −27.12 |
| Ligands | EHOMO | ELUMO | ΔE | η (Chemical Hardness) | μ (Chemical Potential) | ω (Electrophilcity Index) |
|---|---|---|---|---|---|---|
| Arcommunol B | −5.70 | −1.30 | 4.40 | 2.20 | −3.5 | 2.78 |
| Kuwanon S | −5.83 | −1.68 | 4.15 | 2.08 | −3.76 | 3.40 |
| Manuifolin H | −5.19 | −0.01 | 5.18 | 2.59 | −2.60 | 1.31 |
| Kuwanon F | −5.43 | −1.31 | 4.12 | 2.06 | −3.37 | 2.76 |
| Ligands | Lipinski Rule Violation | Veber Rule Violation | PAINS Test | Solubility | BBB | HIA | Acute Oral Toxicity | Carcinogenicity |
|---|---|---|---|---|---|---|---|---|
| Arcommunol B | 0 | 0 | 0 | −4.700 | + | + | 2.347 | - |
| Kuwanon F | 0 | 0 | 0 | −4.078 | + | + | 2.612 | - |
| Kuwanon S | 0 | 0 | 0 | −4.351 | + | + | 2.208 | - |
| Manuifolin H | 0 | 0 | 0 | −3.392 | + | + | 1.8 | - |
| PDB ID | Residues within 5 Å | Native Ligand |
|---|---|---|
| 1V4S | Tyr61, Val62, Arg63, Ser64, Thr65, Pro66, Gln98, Ile159, Met210, Ile211, Tyr214, Tyr215, His218, Cys220, Glu221, Met235, Arg250, Leu451, Val452, Val455, Ala456 | 2-Amino−4-fluoro−5-[(1-methyl−1H-imidazol−2-yl)sulfanyl]-N-(1,3-thiazol−2-yl)benzamide |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faloye, K.O.; Bekono, B.D.; Fakola, E.G.; Ayoola, M.D.; Bello, O.I.; Olajubutu, O.G.; Owoseeni, O.D.; Mahmud, S.; Alqarni, M.; Al Awadh, A.A.; et al. Elucidating the Glucokinase Activating Potentials of Naturally Occurring Prenylated Flavonoids: An Explicit Computational Approach. Molecules 2021, 26, 7211. https://doi.org/10.3390/molecules26237211
Faloye KO, Bekono BD, Fakola EG, Ayoola MD, Bello OI, Olajubutu OG, Owoseeni OD, Mahmud S, Alqarni M, Al Awadh AA, et al. Elucidating the Glucokinase Activating Potentials of Naturally Occurring Prenylated Flavonoids: An Explicit Computational Approach. Molecules. 2021; 26(23):7211. https://doi.org/10.3390/molecules26237211
Chicago/Turabian StyleFaloye, Kolade Olatubosun, Boris Davy Bekono, Emmanuel Gabriel Fakola, Marcus Durojaye Ayoola, Oyenike Idayat Bello, Oluwabukunmi Grace Olajubutu, Onikepe Deborah Owoseeni, Shafi Mahmud, Mohammed Alqarni, Ahmed Abdullah Al Awadh, and et al. 2021. "Elucidating the Glucokinase Activating Potentials of Naturally Occurring Prenylated Flavonoids: An Explicit Computational Approach" Molecules 26, no. 23: 7211. https://doi.org/10.3390/molecules26237211
APA StyleFaloye, K. O., Bekono, B. D., Fakola, E. G., Ayoola, M. D., Bello, O. I., Olajubutu, O. G., Owoseeni, O. D., Mahmud, S., Alqarni, M., Al Awadh, A. A., Alshahrani, M. M., & Obaidullah, A. J. (2021). Elucidating the Glucokinase Activating Potentials of Naturally Occurring Prenylated Flavonoids: An Explicit Computational Approach. Molecules, 26(23), 7211. https://doi.org/10.3390/molecules26237211