1. Introduction
For the excavation of crucial molecular factors on properties and activities, quantitative structure-activity relationship (QSAR) has been an active research area in the past 50+ years. In QSAR, the molecular representations (or descriptors), as the input features of the modeling, represent chemical information of actual entities in computer-understandable numbers [
1,
2,
3]. Historically, molecular fingerprints, such as extended-connectivity fingerprints (ECFPs), have been widely used as representations for the modeling in drug and material discovery [
4]. Recent development in deep neural network facilitates the utilization of different molecular representations such as latent representations [
5,
6]. Latent representations are fixed-length continuous vectors that are derived from autoencoders (AEs) with encoder−decoder architecture [
7]. Although AEs are shown as a generative algorithm for de novo design studies in the beginning [
8,
9], latent representations from encoders of AEs have been extracted for QSAR modeling [
6]. Without sophisticated human-engineered feature selection, these kinds of representations show competitive performance compared with traditional ones [
5]. Furthermore, the representations and QSAR models can be used for multi-objective molecular optimization to tackle inverse QSAR problem [
10].
To obtain latent representations, sequenced-based strings, such as SMILES (Simplified Molecular Input Line Entry Specification), are employed as inputs into AEs. The method in detail is described in the next section. In recent years, great interest has been aroused by developing QSAR models based on latent representations. One of the earliest achievements in this field was the chemical variational autoencoders (VAE) by Aspuru-Guzik et al. [
7]. After generating latent representations for the encoder, two fully connected artificial neural networks (ANNs) were used for the prediction of water−octanol partition coefficient (logP), the synthetic accessibility score, and drug-likeness. More recently, Winter et al. have shown that latent representation applied on support vector machine (SVM) outperforms ECFPs and graph-convolution method by the translation AE model (named CDDD) [
6]. Subsequently, a convolutional neural network (CNN) was trained for the QSAR prediction after generating latent representations [
11,
12]. Owing to the architectural characteristic of local connectivity, shared weight, and pooling [
13], some studies in the literature indicated that CNNs perform better in QSAR modeling compared with ANN and other traditional models [
14].
On the one hand, latent representations should have sequence features if they are derived from sequence-based strings [
15]. As far as we know, recurrent neural network (RNN) outperforms other models including CNN when dealing with sequence data in some areas, such as natural language processing (NLP) [
16] and electrocardiogram classification (ECG) [
17]. Even though CNN performs well due to its ability of local feature selection, RNN has its advantage in global feature discovery [
18]. With regard to molecules, it has similarity with the NLP and ECG. We assume that the local feature represents the type of atoms and functional group, and global feature is the atomic arrangement. Molecular properties and activities depend on not only atom types and functional groups (local features) but also the arrangement (global features). In other words, the sequence of the atoms or functional groups plays an important role in molecular properties. However, very few works have been done to study the modeling performance with the difference of the molecular sequence (global features).
On the other hand, the scarce availability of labeled data is the major obstacle for QSAR modeling [
2]. According to the probably approximately correct theory, the size of training data plays a key role in the accuracy of machine learning methods [
19]. Nonetheless, the available datasets are small at most stages of the QSAR pipeline, especially bioactivity modeling. One strategy to solve this problem is transfer learning algorithms [
20]. Transfer learning is one kind of machine learning method that takes advantage of existing, generalizable knowledge from other sources [
21]. For example, Li et al. reported an RNN model pretrained on one million unlabeled molecules from ChEMBL and fine-tuned with Lipophilicity (LogP), HIV, and FreeSolv data [
22]. It indicated that transfer learning improved the performance strongly compared with learning from scratch. Additionally, Iovanac et al. improve the QSAR prediction ability by the integration of experimentally available pKa data and DFT-based characterizations of the (de)protonation free energy [
23]. However, few transfer learning strategies for multiple bioactivity and material datasets have been presented. In comparison with physicochemical and physiological properties, the measurements of molecular bioactivities are more time and resource consuming. Additionally, the scales of the datasets about experimental material properties are usually less than 1000 [
24]. Therefore, it is advisable to train more transfer learning models among this type of dataset. In this way, the information from other datasets would be transferred into small ones to facilitate molecular design and drug discovery.
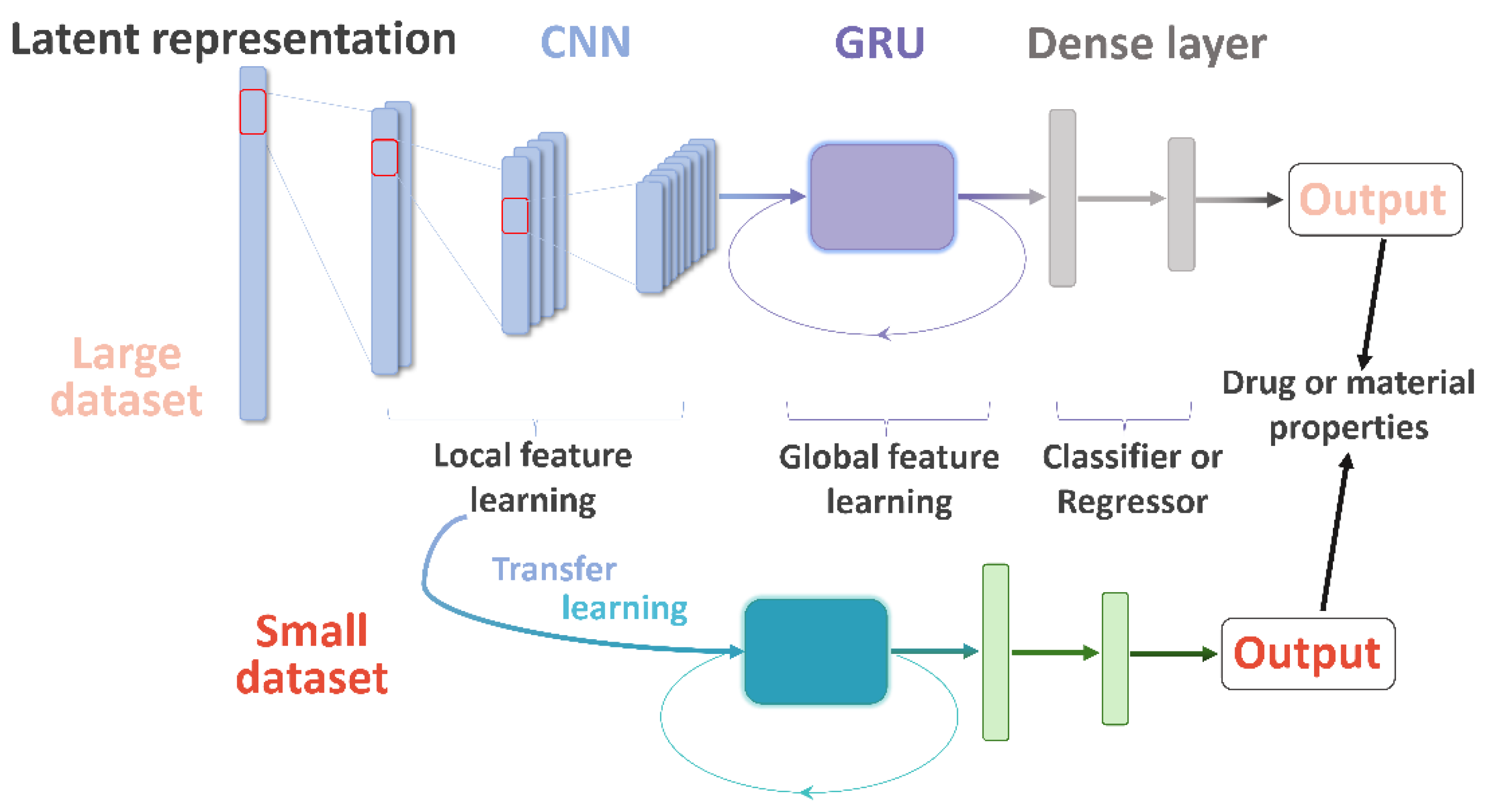
Herein, we describe the convolutional recurrent neural network and transfer learning (CRNNTL) method to tackle the problems with molecular sequence and data scarcity. The convolutional recurrent neural network (CRNN) is loosely inspired by the architectures proposed in the applications of polyphonic sound detection and electrocardiogram classification [
25,
26,
27], which integrates the advantages of CNN and RNN (gated recurrent units used here, named GRU) at the same time. The concept is illustrated in
Figure 1 in which the model is constituted by the CNN, GRU, and dense layer parts. Firstly, the CRNN model is tested using diverse benchmark datasets including drug and material properties and compared with CNN and classical methods, such as random forest (RF) and SVM. Then, an isomers-based dataset is trained by CRNN and CNN to elucidate the improved ability of CRNN in the global feature learning. Next, we demonstrated that the transfer learning part of CRNNTL could be used to improve the performance for scarce bioactivity and material datasets. Finally, the high versatility of CRNNTL was shown by QSAR modeling based on two other latent representations derived from different types of AEs.
4. Conclusions
We demonstrate a convolutional and recurrent neural network and transfer learning model (CRNNTL) for both QSAR modeling and knowledge transfer study. As for the CRNN part, it integrates the advantages of both convolutional and recurrent neural networks as well as the data augmentation method. Our method outperforms or competes with the state-of-art ones in 27 datasets of drug or material properties. We hypothesize that the excellent performance results from improvement of the ability of global feature (atomic arrangement) extraction by the GRU part. Meanwhile, the ability of local feature (types of atoms and functional groups) extraction is maintained by the CNN part. This assumption was strengthened by training and testing isomer-based datasets. Even though more parameters need to be trained when the data size is not large, the performance of CRNN is almost 10% higher than that of traditional CNN because of the outstanding ability of global feature learning. In drug and material discovery, high-proportioned isomers and derivatives need to be tested for their properties. Therefore, our model provides a new strategy for molecular QSAR modeling in various properties.
With regard to the transfer learning part, effective knowledge transfer from a larger dataset into a small one can occur for QSAR modeling in both drug and material properties. When considering binding site similarities, up to 30% improvement was achieved by CRNNTL between different bioactivity datasets. In addition, a considerable increase was shown for the transfer learning between the larger dataset for luminescent molecules and the smaller one for OLED materials. Accordingly, transfer learning could facilitate the QSAR performance when considering the correlation between datasets. Hence, CRNNTL could be a potential method to overcome the data scarcity for QSAR modeling.
At last, CRNNTL showed high versatility by testing the model on different latent representations from other types of autoencoders. We anticipate that the performance of QSAR modeling by our method could be further improved by the latent representation generated from the AEs, which is more suitable for molecules in material science. Accordingly, we expect that CRNNTL could pave the pathway for drug and material discovery in the big data era.






{kind=link}
{kind=link}





