
Innate Immunity Modulating Impurities and the Immunotoxicity of Nanobiotechnology-Based Drug Products



Abstract
:1. Introduction
2. Innate Immunity Modulating Impurities
3. Impact of IIMIs on the Immunotoxicity of Drug Products
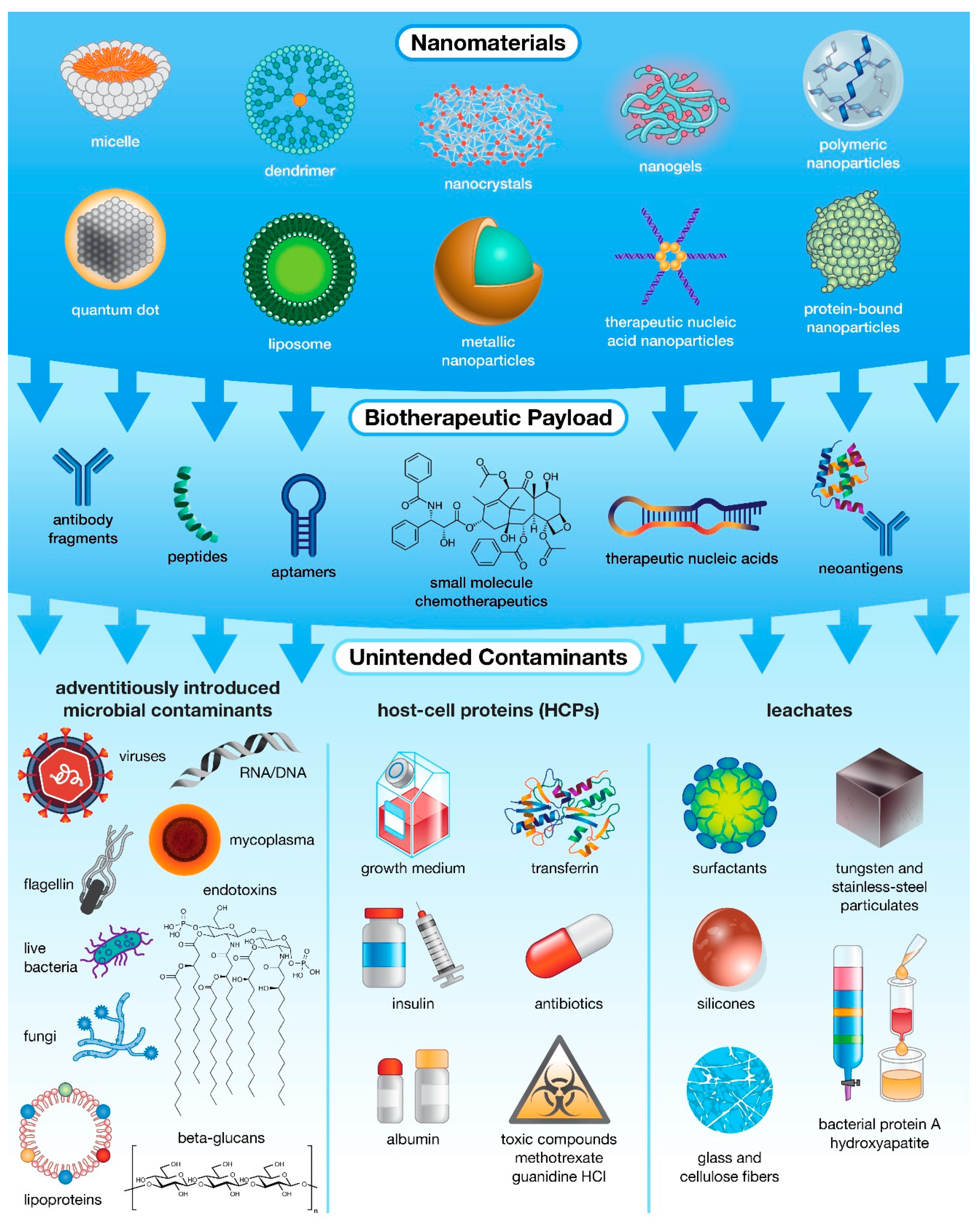
4. Sources of Immunotoxicity in Nanotechnology-Based Products
5. IIMIs Commonly Found in Pharmaceutical Products
5.1. Microbial Components
5.2. Whole Microbes
5.3. Leachates
5.4. Host Cell Proteins
6. Immune-Mediated Adverse Effects to Pharmaceutical Products
7. Methods for IIMI Detection
7.1. Direct Detection Methods
7.2. Indirect Detection Methods
7.3. Biological Staining and Gel-Based Methods
7.4. Antibody-Based Enzymatic Methods
7.5. Nucleic Acid Hybridization Methods
7.6. Cell-Based Methods
7.7. In Vivo Methods
8. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Elsabahy, M.; Wooley, K.L. Cytokines as biomarkers of nanoparticle immunotoxicity. Chem. Soc. Rev. 2013, 42, 5552–5576. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Mehrotra, S.; Agarwal, S. The paradigm of Th1 and Th2 cytokines. Immunol. Res. 1999, 20, 147–161. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.; van der Meer, J.W.; Mhlanga, M.M.; Mulder, W.J. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrantes-Metz, R.M.; Adams, C.; Metz, A. Pharmaceutical development phases: A duration analysis. FTC Bur. Econ. Work. Pap. 2004, 274, 30. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Kaitin, K.I.; DiMasi, J.A. Pharmaceutical innovation in the 21st century: New drug approvals in the first decade, 2000–2009. Clin. Pharmacol. Ther. 2011, 89, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Verthelyi, D.; Wang, V. Trace levels of innate immune response modulating impurities (IIRMIs) synergize to break tolerance to therapeutic proteins. PLoS ONE 2010, 5, e15252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiPaolo, B.; Pennetti, A.; Nugent, L.; Venkat, K. Monitoring impurities in biopharmaceuticals produced by recombinant technology. Pharm. Sci. Technol. Today 1999, 2, 70–82. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A. Pre-clinical immunotoxicity studies of nanotechnology-formulated drugs: Challenges, considerations and strategy. J. Control. Release 2015, 220, 571–583. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Shurin, M.; Shvedova, A.A. Current understanding of interactions between nanoparticles and the immune system. Toxicol. Appl. Pharm. 2016, 299, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadpour, R.; Ghandehari, H. Mechanisms of Immune Response to Inorganic Nanoparticles and Their Degradation Products. Adv. Drug Deliv. Rev. 2021, 180, 114022. [Google Scholar] [CrossRef] [PubMed]
- Reijers, J.A.A.; Malone, K.E.; Bajramovic, J.J.; Verbeek, R.; Burggraaf, J.; Moerland, M. Adverse immunostimulation caused by impurities: The dark side of biopharmaceuticals. Br. J. Clin. Pharm. 2019, 85, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.S.; Sauna, Z.E. Immunogenicity assessment during the development of protein therapeutics. J. Pharm Pharmcol. 2018, 70, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Sauna, Z.E.; Lagassé, D.; Pedras-Vasconcelos, J.; Golding, B.; Rosenberg, A.S. Evaluating and Mitigating the Immunogenicity of Therapeutic Proteins. Trends Biotechnol. 2018, 36, 1068–1084. [Google Scholar] [CrossRef] [PubMed]
- Sauna, Z.E.; Richards, S.M.; Maillere, B.; Jury, E.C.; Rosenberg, A.S. Editorial: Immunogenicity of Proteins Used as Therapeutics. Front. Immunol. 2020, 11, 614856. [Google Scholar] [CrossRef]
- Kessler, M.; Goldsmith, D.; Schellekens, H. Immunogenicity of biopharmaceuticals. Nephrol. Dial. Transplant. 2006, 21, v9–v12. [Google Scholar] [CrossRef] [PubMed]
- Jawa, V.; Joubert, M.K.; Zhang, Q.; Deshpande, M.; Hapuarachchi, S.; Hall, M.P.; Flynn, G.C. Evaluating immunogenicity risk due to host cell protein impurities in antibody-based biotherapeutics. AAPS J. 2016, 18, 1439–1452. [Google Scholar] [CrossRef]
- Wang, X.; Hunter, A.K.; Mozier, N.M. Host cell proteins in biologics development: Identification, quantitation and risk assessment. Biotechnol. Bioeng. 2009, 103, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Ilinskaya, A.N.; Dobrovolskaia, M.A. Understanding the immunogenicity and antigenicity of nanomaterials: Past, present and future. Toxicol. Appl. Pharmacol. 2016, 299, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Jawa, V.; Cousens, L.P.; Awwad, M.; Wakshull, E.; Kropshofer, H.; De Groot, A.S. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clin. Immunol. 2013, 149, 534–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, P. The Immune System, 3rd ed.; Garland Science, Taylor & Francis Group, LLC: New York, NY, USA, 2009. [Google Scholar]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Creagh, E.M.; O’Neill, L.A. TLRs, NLRs and RLRs: A trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006, 27, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Kobiyama, K.; Takeshita, F.; Ishii, K.J. Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front. Cell. Infect. Microbiol. 2013, 2, 168. [Google Scholar] [CrossRef] [Green Version]
- Gorden, K.B.; Gorski, K.S.; Gibson, S.J.; Kedl, R.M.; Kieper, W.C.; Qiu, X.; Tomai, M.A.; Alkan, S.S.; Vasilakos, J.P. Synthetic TLR agonists reveal functional differences between human TLR7 and TLR8. J. Immunol. 2005, 174, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Technical Committee ISO/TC 229, N. ISO 29701:2010(en) Nanotechnologies—Endotoxin Test on Nanomaterial Samples for In Vitro systems—Limulus Amebocyte Lysate (LAL) Test. Available online: https://www.iso.org/obp/ui/es/#iso:std:iso:29701:ed-1:v1:en (accessed on 17 November 2021).
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Haile, L.A.; Puig, M.; Polumuri, S.K.; Ascher, J.; Verthelyi, D. In vivo effect of innate immune response modulating impurities on the skin milieu using a macaque model: Impact on product immunogenicity. J. Pharm. Sci. 2017, 106, 751–760. [Google Scholar] [CrossRef] [Green Version]
- Medvedev, A.E.; Sabroe, I.; Hasday, J.D.; Vogel, S.N. Invited review: Tolerance to microbial TLR ligands: Molecular mechanisms and relevance to disease. J. Endotoxin Res. 2006, 12, 133–150. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Wolf, A.J.; Underhill, D.M. β-glucan recognition by the innate immune system. Immunol. Rev. 2009, 230, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Center for Devices and Radiological Health, U.S. FOOD & DRUG. Immunotoxicity Testing Guidance. 1999; pp. 1–16. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunotoxicity-testing-guidance (accessed on 17 August 2021).
- Descotes, J. Methods of evaluating immunotoxicity. Expert Opin. Drug Metab. Toxicol. 2006, 2, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.K.; Lichtman, A.H.; Pillai, S. Hypersensitivity Disorders. In Cellular and Molecular Immunology E-book, 8th ed.; Elsevier Health Sciences: Philadelphia, PA, USA, 2014. [Google Scholar]
- Rosenberg, A. Immunogenicity of biological therapeutics: A hierarchy of concerns. Dev. Biol. 2003, 112, 15–22. [Google Scholar]
- Vial, T.; Choquet-Kastylevsky, G.; Descotes, J. Adverse effects of immunotherapeutics involving the immune system. Toxicology 2002, 174, 3–11. [Google Scholar] [CrossRef]
- Gell, P.G.H.; Coombs, R.R.A. The Classification of Allergic Reactions Underlying Disease. In Clinical Aspects of Immunology; Blackwell: Oxford, UK, 1963; pp. 317–337. [Google Scholar]
- Krishna, M.; Nadler, S.G. Immunogenicity to biotherapeutics–the role of anti-drug immune complexes. Front. Immunol. 2016, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szebeni, J.; Simberg, D.; González-Fernández, Á.; Barenholz, Y.; Dobrovolskaia, M.A. Roadmap and strategy for overcoming infusion reactions to nanomedicines. Nat. Nanotechnol. 2018, 13, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Sarma, J.V.; Ward, P.A. The complement system. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: An innate response to nanomedicines acting as pseudo-viruses. Eur. J. Nanomed. 2015, 7, 203–205. [Google Scholar] [CrossRef]
- Finkelman, F.D.; Khodoun, M.V.; Strait, R. Human IgE-independent systemic anaphylaxis. J. Allergy Clin. Immunol. 2016, 137, 1674–1680. [Google Scholar] [CrossRef] [Green Version]
- Ali, H. Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol. Lett. 2010, 128, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Fennrich, S.; Hennig, U.; Toliashvili, L.; Schlensak, C.; Wendel, H.P.; Stoppelkamp, S. More than 70 years of pyrogen detection: Current state and future perspectives. Altern. Lab. Anim. 2016, 44, 239–253. [Google Scholar] [CrossRef]
- Binkowska, A.M.; Michalak, G.; Pilip, S.; Kopacz, M.; Słotwiński, R. The diagnostic value of early cytokine response in patients after major trauma-preliminary report. Cent. Eur. J. Immunol. 2018, 43, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Administration, U.S.F. a. D. Inspection Technical Guides: Bacterial Endotoxins/Pyrogens. Health and Human Services, D. a. D. P. Ed. 1985; pp. 1–9. Available online: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-technical-guides/bacterial-endotoxinspyrogens (accessed on 4 October 2021).
- Dobrovolskaia, M.A.; McNeil, S.E. Strategy for selecting nanotechnology carriers to overcome immunological and hematological toxicities challenging clinical translation of nucleic acid-based therapeutics. Expert Opin. Drug Deliv. 2015, 12, 1163–1175. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Kawabata, K.; Sakurai, F.; Nakagawa, S.; Mizuguchi, H. Innate immune response induced by gene delivery vectors. Int. J. Pharm. 2008, 354, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Shvedova, A.A.; Kisin, E.R.; Mercer, R.; Murray, A.R.; Johnson, V.J.; Potapovich, A.I.; Tyurina, Y.Y.; Gorelik, O.; Arepalli, S.; Schwegler-Berry, D. Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2005, 289, L698–L708. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-Y.; Lu, S.-L.; Hu, C.-W.; Yeh, C.-S.; Lee, G.-B.; Lei, H.-Y. Size-dependent attenuation of TLR9 signaling by gold nanoparticles in macrophages. J. Immunol. 2012, 188, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Crist, R.M.; Grossman, J.H.; Patri, A.K.; Stern, S.T.; Dobrovolskaia, M.A.; Adiseshaiah, P.P.; Clogston, J.D.; McNeil, S.E. Common pitfalls in nanotechnology: Lessons learned from NCI’s Nanotechnology Characterization Laboratory. Integr. Biol. (Camb.) 2013, 5, 66–73. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A. Nucleic Acid Nanoparticles at a Crossroads of Vaccines and Immunotherapies. Molecules 2019, 24, 4620. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Germolec, D.R.; Weaver, J.L. Evaluation of nanoparticle immunotoxicity. Nat. Nanotechnol. 2009, 4, 411–414. [Google Scholar] [CrossRef]
- Shah, A.; Dobrovolskaia, M.A. Immunological effects of iron oxide nanoparticles and iron-based complex drug formulations: Therapeutic benefits, toxicity, mechanistic insights, and translational considerations. Nanomedicine 2018, 14, 977–990. [Google Scholar] [CrossRef]
- Zolnik, B.S.; González-Fernández, A.; Sadrieh, N.; Dobrovolskaia, M.A. Nanoparticles and the immune system. Endocrinology 2010, 151, 458–465. [Google Scholar] [CrossRef]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Does size matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef]
- Simak, J.; De Paoli, S. The effects of nanomaterials on blood coagulation in hemostasis and thrombosis. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1448. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Patri, A.K.; Simak, J.; Hall, J.B.; Semberova, J.; De Paoli Lacerda, S.H.; McNeil, S.E. Nanoparticle size and surface charge determine effects of PAMAM dendrimers on human platelets in vitro. Mol. Pharm. 2012, 9, 382–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center for Drug Evaluation and Research, U.S. FOOD & DRUG. Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products. 2014; Volume FDA-2013-D-0092, pp. 1–36. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunogenicity-assessment-therapeutic-protein-products (accessed on 4 October 2021).
- 〈85〉 Bacterial Endotoxins Test. In USP 35-NF 30, (USP-NF). Available online: http://www.houshiji.com/Uploads/Download/5a27396519552.pdf (accessed on 17 November 2021).
- 〈151〉 Pyrogen Test. In USP 40-NF 35, (USP-NF). Available online: http://www.uspbpep.com/usp31/v31261/usp31nf26s1_c151.asp (accessed on 17 November 2021).
- Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-pyrogen-and-endotoxins-testing-questions-and-answers (accessed on 17 November 2021).
- Center for Devices and Radiological Health, U.S. FOOD & DRUG. Endotoxin Testing Recommendations for Single-Use Intraocular Ophthalmic Devices. 2015. Available online: https://www.fda.gov/media/88615/download (accessed on 17 November 2021).
- Questions and Answers on Quality Related Controlled Correspondence. 2021. Available online: https://www.fda.gov/media/152281/download (accessed on 17 November 2021).
- Biological Evaluation of Medical Devices–Part 1: Evaluation and Testing within a Risk Management Process. Available online: https://www.iso.org/obp/ui/#iso:std:iso:10993:-1:ed-5:v2:en (accessed on 17 November 2021).
- Haile, L.A.; Puig, M.; Kelley-Baker, L.; Verthelyi, D. Detection of innate immune response modulating impurities in therapeutic proteins. PLoS ONE 2015, 10, e0125078. [Google Scholar]
- Wadhwa, M.; Knezevic, I.; Kang, H.-N.; Thorpe, R. Immunogenicity assessment of biotherapeutic products: An overview of assays and their utility. Biologicals 2015, 43, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Eaton, L.C. Host cell contaminant protein assay development for recombinant biopharmaceuticals. J. Chromatogr. A 1995, 705, 105–114. [Google Scholar] [CrossRef]
- Pyrogenicit–Principles and Methods for Pyrogen Testing of Medical Devices. Available online: https://www.iso.org/obp/ui/#iso:std:iso:tr:21582:ed-1:v1:en (accessed on 17 November 2021).
- Singh, S.K.; Afonina, N.; Awwad, M.; Bechtold-Peters, K.; Blue, J.T.; Chou, D.; Cromwell, M.; Krause, H.-J.; Mahler, H.-C.; Meyer, B.K.; et al. An Industry Perspective on the Monitoring of Subvisible Particles as a Quality Attribute for Protein Therapeutics. J. Pharm. Sci. 2010, 99, 3302–3321. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.; Cherney, B.; Lubinecki, A.; Ma, S.; Marszal, E.; Mire-Sluis, A.; Nikolai, T.; Novak, J.; Ragheb, J.; Simak, J. Meeting report on protein particles and immunogenicity of therapeutic proteins: Filling in the gaps in risk evaluation and mitigation. Biologicals 2010, 38, 602–611. [Google Scholar] [CrossRef]
- Pacáková, V.; Suchánková, J.; Štulík, K. Separation of biologically active peptides by capillary electrophoresis and high-performance liquid chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1996, 681, 69–76. [Google Scholar] [CrossRef]
- Baumann, A. Early development of therapeutic biologics-pharmacokinetics. Curr. Drug Metab. 2006, 7, 15–21. [Google Scholar] [CrossRef]
- Liu, Y.; Romijn, E.P.; Verniest, G.; Laukens, K.; De Vijlder, T. Mass spectrometry-based structure elucidation of small molecule impurities and degradation products in pharmaceutical development. TrAC Trends Anal. Chem. 2019, 121, 115686. [Google Scholar] [CrossRef]
- Barbosa, M.D.; Kumar, S.; Loughrey, H.; Singh, S.K. Biosimilars and biobetters as tools for understanding and mitigating the immunogenicity of biotherapeutics. Drug Discov. Today 2012, 17, 1282–1288. [Google Scholar] [CrossRef]
- Jawa, V.; Terry, F.; Gokemeijer, J.; Mitra-Kaushik, S.; Roberts, B.J.; Tourdot, S.; De Groot, A.S. T-cell dependent immunogenicity of protein therapeutics pre-clinical assessment and mitigation–updated consensus and review 2020. Front. Immunol. 2020, 11, 1301. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Salimi-Moosavi, H. Bioanalysis of target biomarker and PK/PD relevancy during the development of biotherapeutics. Bioanalysis 2012, 4, 2513–2523. [Google Scholar] [CrossRef]
- Wang, J.; Lozier, J.; Johnson, G.; Kirshner, S.; Verthelyi, D.; Pariser, A.; Shores, E.; Rosenberg, A. Neutralizing antibodies to therapeutic enzymes: Considerations for testing, prevention and treatment. Nat. Biotechnol. 2008, 26, 901–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Regenmortel, M.H. Antigenicity and immunogenicity of synthetic peptides. Biologicals 2001, 29, 209–213. [Google Scholar] [CrossRef]
- Group, B.D.W.; Atkinson Jr, A.J.; Colburn, W.A.; DeGruttola, V.G.; DeMets, D.L.; Downing, G.J.; Hoth, D.F.; Oates, J.A.; Peck, C.C.; Schooley, R.T. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- Wagner, J.A. Strategic approach to fit-for-purpose biomarkers in drug development. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 631–651. [Google Scholar] [CrossRef]
- Fisher, A.C.; Lee, S.L.; Harris, D.P.; Buhse, L.; Kozlowski, S.; Yu, L.; Kopcha, M.; Woodcock, J. Advancing pharmaceutical quality: An overview of science and research in the US FDA’s Office of Pharmaceutical Quality. Int. J. Pharm. 2016, 515, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Neun, B.W.; Dobrovolskaia, M.A. NCL Method STE-1.2: Detection and Quantification of Gram Negative Bacterial Endotoxin Contamination in Nanoparticle Formulations by Kinetic Turbidity LAL Assay. Available online: https://ncl.cancer.gov/sites/default/files/protocols/NCL_Method_STE-1.2.pdf (accessed on 24 August 2021).
- Nakagawa, Y.; Maeda, H.; Murai, T. Evaluation of the in vitro pyrogen test system based on proinflammatory cytokine release from human monocytes: Comparison with a human whole blood culture test system and with the rabbit pyrogen test. Clin. Diagn. Lab. Immunol. 2002, 9, 588–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikkert, R.; de Groot, E.R.; Aarden, L.A. Cytokine induction by pyrogens: Comparison of whole blood, mononuclear cells, and TLR-transfectants. J. Immunol. Methods 2008, 336, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Neun, B.W.; Cedrone, E.; Potter, T.M.; Crist, R.M.; Dobrovolskaia, M.A. Detection of Beta-Glucan Contamination in Nanotechnology-Based Formulations. Molecules 2020, 25, 3367. [Google Scholar] [CrossRef]
- Neun, B.W.; Dobrovolskaia, M.A. NCL Method STE-4: Detection and Quantification of β-(1,3)-D-Glucan Contamination in Nanoparticle Formulations by Factor C Depleted LAL (Glucatell®) Assay. Available online: https://ncl.cancer.gov/sites/default/files/NCL_Method_STE-4.pdf (accessed on 24 August 2021).
- Dobrovolskaia, M.A.; Neun, B.W.; Clogston, J.D.; Ding, H.; Ljubimova, J.; McNeil, S.E. Ambiguities in applying traditional Limulus amebocyte lysate tests to quantify endotoxin in nanoparticle formulations. Nanomedicine 2010, 5, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Dobrovolskaia, M.A.; Neun, B.W.; Clogston, J.D.; Grossman, J.H.; McNeil, S.E. Choice of method for endotoxin detection depends on nanoformulation. Nanomedicine 2014, 9, 1847–1856. [Google Scholar] [CrossRef]
- Neun, B.W.; Dobrovolskaia, M.A. Detection and quantitative evaluation of endotoxin contamination in nanoparticle formulations by LAL-based assays. Methods Mol. Biol. 2011, 697, 121–130. [Google Scholar]
- Neun, B.W.; Dobrovolskaia, M.A. Detection of endotoxin in nano-formulations using limulus amoebocyte lysate (LAL) assays. JoVE (J. Vis. Exp.) 2019, 143, e58830. [Google Scholar] [CrossRef]
- Petersen, E.J.; Henry, T.B.; Zhao, J.; MacCuspie, R.I.; Kirschling, T.L.; Dobrovolskaia, M.A.; Hackley, V.; Xing, B.; White, J.C. Identification and avoidance of potential artifacts and misinterpretations in nanomaterial ecotoxicity measurements. Environ. Sci. Technol. 2014, 48, 4226–4246. [Google Scholar] [CrossRef]
- Gunn, G.R.; Evans, C.; Yang, E. Immunogenicity and biomarkers: Bioanalytical challenges and considerations. Future Sci. 2017, 9, 1729–1732. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, M.; Thorpe, R. Harmonization and standardization of immunogenicity assessment of biotherapeutic products. Bioanalysis 2019, 11, 1593–1604. [Google Scholar] [CrossRef] [Green Version]
- Forster, R.J.; Bertoncello, P.; Keyes, T.E. Electrogenerated chemiluminescence. Annu. Rev. Anal. Chem. 2009, 2, 359–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolter, T.; Richter, A. Assays for controlling host-cell impurities in biopharmaceuticals. BioProcess Int. 2005, 3, 40–46. [Google Scholar]
- Neun, B.W.; Cedrone, E.; Dobrovolskaia, M.A. NCL Method STE 5.2: Analysis of Complement Activation by Single-Plex EIA or Multiplex ELISA. Available online: https://ncl.cancer.gov/sites/default/files/protocols/NCL_Method_ITA-5.2.pdf (accessed on 24 August 2021).
- Potter, T.M.; Cedrone, E.; Neun, B.W.; Dobrovolskaia, M.A. NCL Method-10: Preparation of Human Whole Blood and Peripheral Blood Mononuclear Cell Cultures to Analyze Nanoparticle Potential to Induce Cytokines In Vitro. Available online: https://ncl.cancer.gov/sites/default/files/NCL_Method_ITA-10.pdf (accessed on 24 August 2021).
- Dill, K. Sensitive analyte detection and quantitation using the threshold immunoassay system. ACS Publ. 1996, 9, 89–102. [Google Scholar]
- Mufarrege, E.F.; Haile, L.A.; Etcheverrigaray, M.; Verthelyi, D.I. Multiplexed gene expression as a characterization of bioactivity for interferon beta (IFN-β) biosimilar candidates: Impact of innate immune response modulating impurities (IIRMIs). AAPS J. 2019, 21, 26. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Document | Type | Purpose | Reference |
|---|---|---|---|
| USP 85 Bacterial Endotoxins Test | GI | Describes method validation and sample preparation requirements for turbidity, chromogenic and gel-clot LAL assay | [63] |
| USP 151 Pyrogen Test | GI | Describes method validation and sample preparation requirements for the rabbit pyrogen test | [64] |
| FDA Immunotoxicity Testing Guidance (FDA-modified version of ISO-10993) | GI | Summarizes general types of toxicity and subsequent testing that should be considered for medical devices or constituent materials | [35] |
| FDA Guidance for Industry: Pyrogen and Endotoxin Testing: Questions and Answers | GI | Provides bacterial endotoxin and pyrogen testing recommendations (gel-blot, photometric, and kinetic tests) and acceptance criteria | [65] |
| FDA Endotoxin Testing Recommendations for Single-Use Intraocular Ophthalmic Devices | GI | Provides recommended endotoxin limits for the release of intraocular devices and single-use intraocular ophthalmic surgical instruments/accessories | [66] |
| FDA Questions and Answers on Quality Related Controlled Correspondence | GI | Provides answers to common scientific and regulatory questions around the manufacture and quality control of generic drug production including endotoxin testing | [67] |
| FDA Immunogenicity Assessment for Therapeutic Protein Products | GI | Outlines approaches to evaluate and mitigate adverse immune responses/immunogenicity associated with therapeutic protein products; discusses the importance of IIMI detection | [62] |
| ISO-10993-1 Biological Evaluation and Testing Standards for Medical Devices (prepared by ISO/TC 149) | IS | Outlines the potential biological risks arising from the use of medical devices and provides a framework to plan biological evaluation, testing methods, and acceptance criteria | [68] |
| ISO-29701 Endotoxin Standard (prepared by ISO/TC 229) | IS | Describes application of LAL assay for evaluation of endotoxin levels in nanomaterials intended for use in vitro | [29] |
| ISO-21582 Pyrogenicity Standard (prepared by ISO/TC 149) | IS | Specifies the principles and methods for pyrogen testing of medical devices and their materials | [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holley, C.K.; Dobrovolskaia, M.A. Innate Immunity Modulating Impurities and the Immunotoxicity of Nanobiotechnology-Based Drug Products. Molecules 2021, 26, 7308. https://doi.org/10.3390/molecules26237308
Holley CK, Dobrovolskaia MA. Innate Immunity Modulating Impurities and the Immunotoxicity of Nanobiotechnology-Based Drug Products. Molecules. 2021; 26(23):7308. https://doi.org/10.3390/molecules26237308
Chicago/Turabian StyleHolley, Claire K., and Marina A. Dobrovolskaia. 2021. "Innate Immunity Modulating Impurities and the Immunotoxicity of Nanobiotechnology-Based Drug Products" Molecules 26, no. 23: 7308. https://doi.org/10.3390/molecules26237308
APA StyleHolley, C. K., & Dobrovolskaia, M. A. (2021). Innate Immunity Modulating Impurities and the Immunotoxicity of Nanobiotechnology-Based Drug Products. Molecules, 26(23), 7308. https://doi.org/10.3390/molecules26237308