
A Computational Journey across Nitroxide Radicals: From Structure to Spectroscopic Properties and Beyond



Abstract
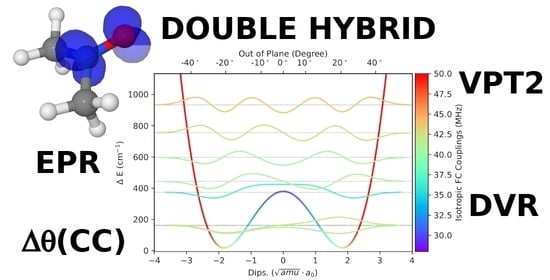
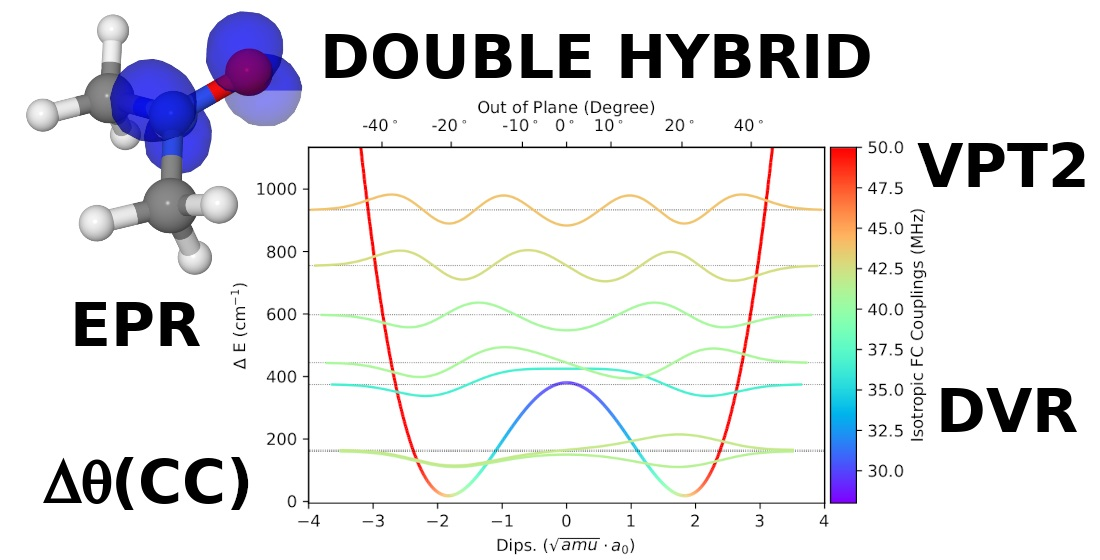
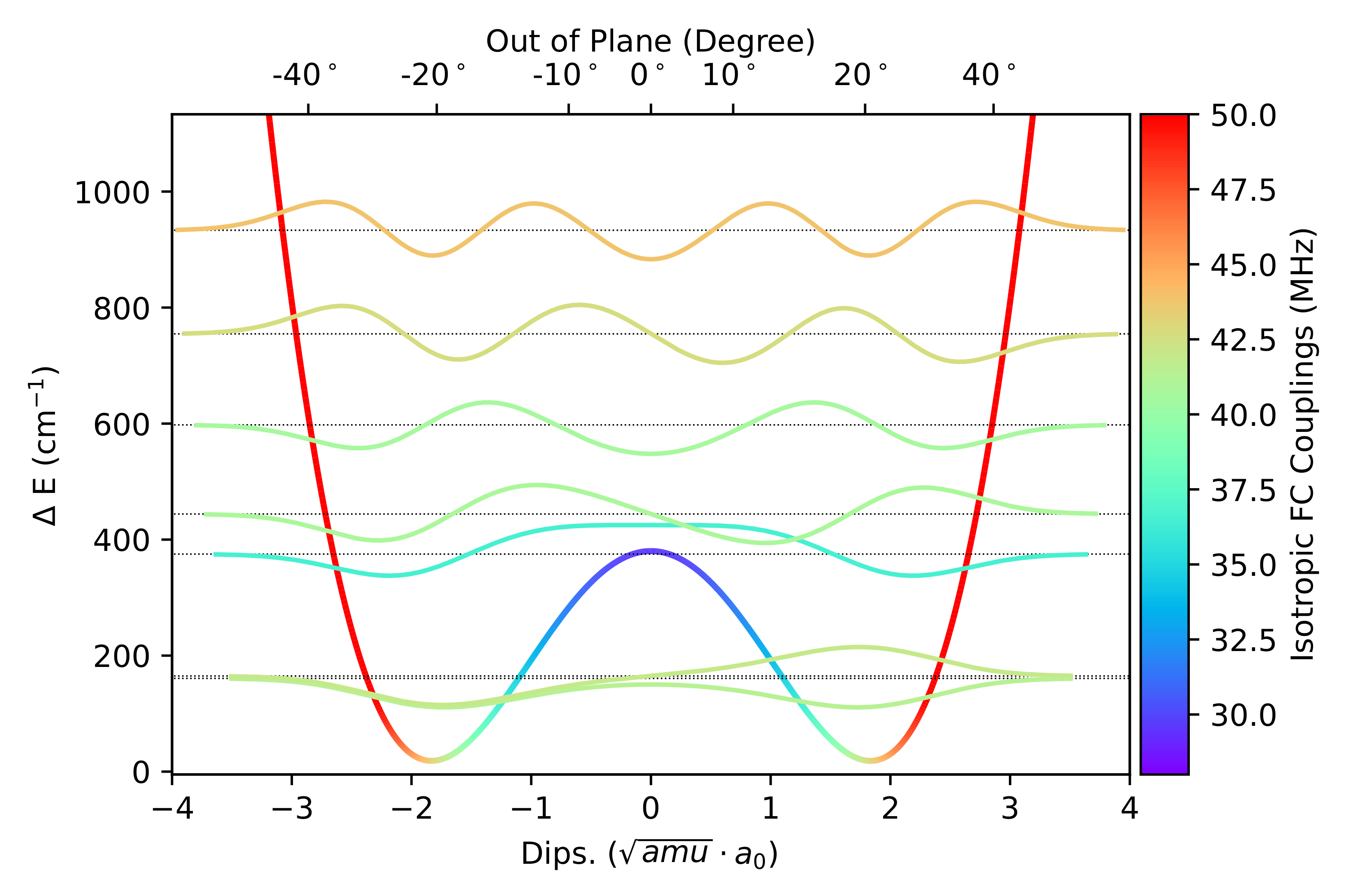
:
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Template Molecule
3.2. Geometries
3.3. NO Stretching Frequencies
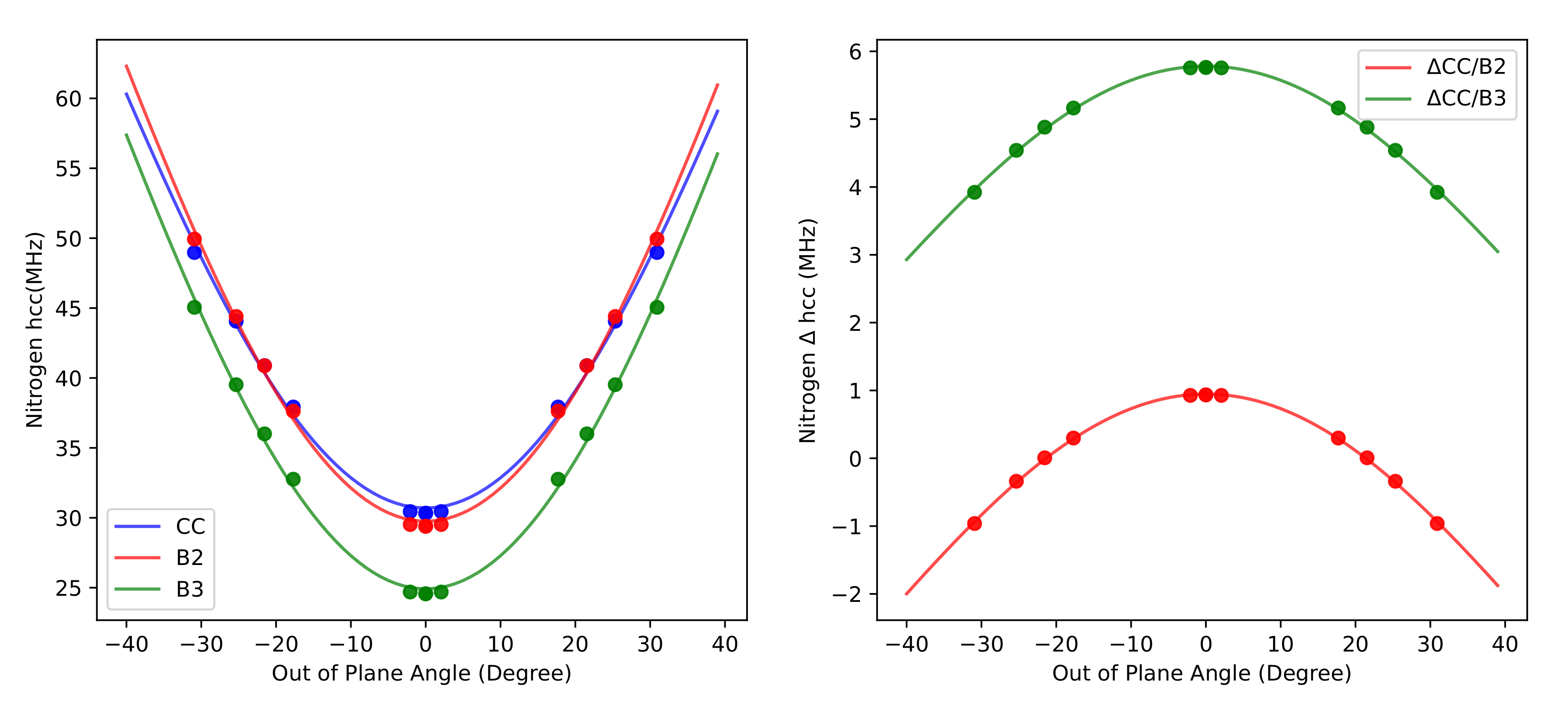
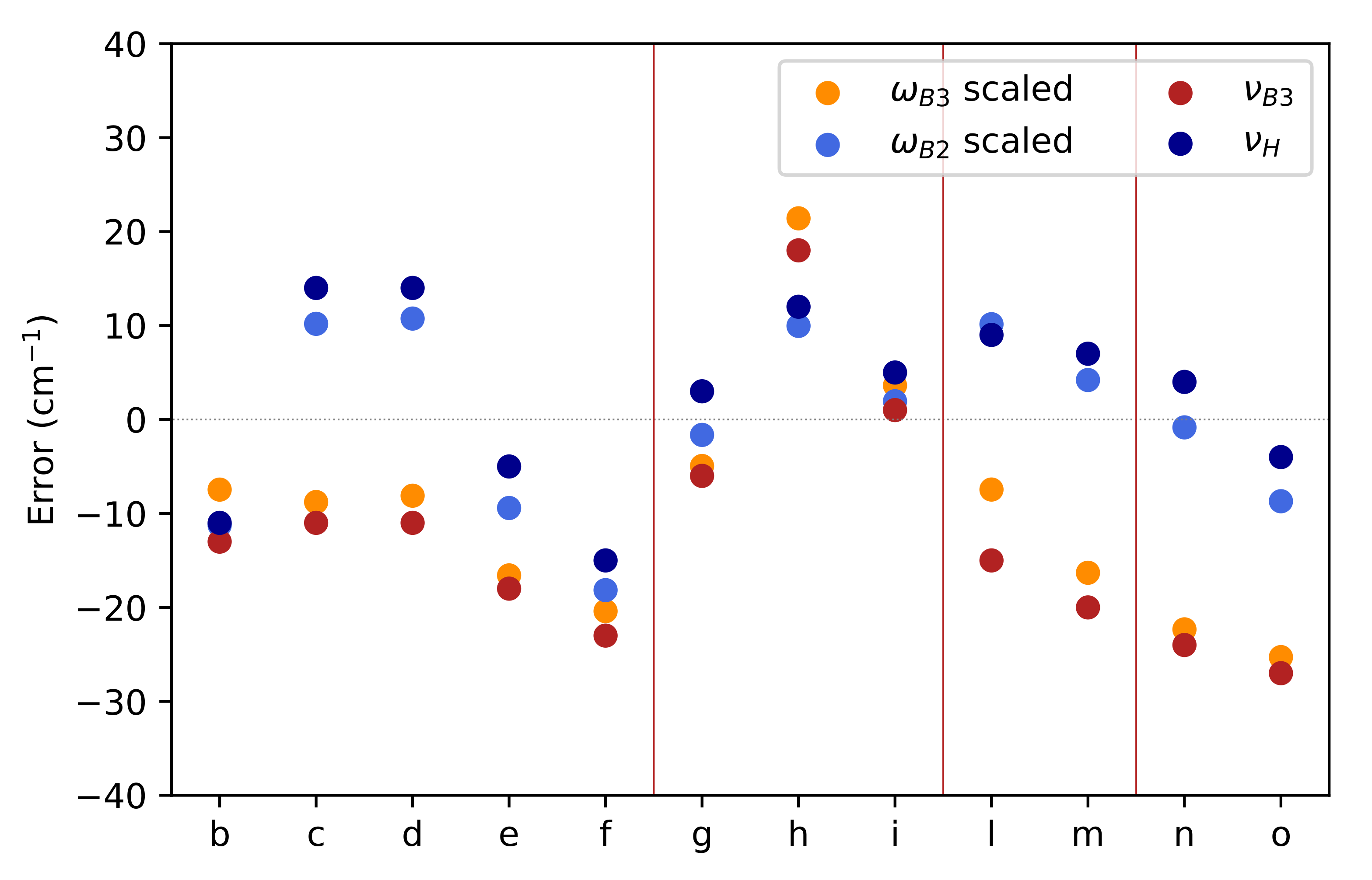
3.4. Hyperfine Coupling Constants
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berliner, L.J. Spin Labeling: Theory and Applications; Academic Press: New York, NY, USA, 1976. [Google Scholar]
- Kocherginsky, N.; Swantz, H.M. Nitroxide Spin Labels: Reactions in Biology and Chemistry; CRC Press: New York, NY, USA, 1995. [Google Scholar]
- Buchaklian, A.H.; Klug, C.S. Characterization of the walker a motif of MsbA using site-directed spin labeling electron paramagnetic resonance spectroscopy. Biochemistry 2005, 44, 5503–5509. [Google Scholar] [CrossRef]
- Giovannini, T.; Lafiosca, P.; Balasubramanian, C.; Barone, V.; Cappelli, C. Effective yet reliable computation of hyperfine coupling constants in solution by a QM/MM approach: Interplay between electrostatic and non-electrostatic effects. J. Chem. Phys. 2019, 150, 124102. [Google Scholar] [CrossRef]
- Sharma, B.; Tran, V.A.; Pongratz, T.; Galazzo, L.; Zhurko, I.; Bordignon, E.; Kast, S.M.; Neese, F.; Marx, D. A joint venture of ab initio molecular dynamics, coupled cluster electronic structure methods, and liquid state theory to compute accurate isotropic hyperfine constants of nitroxide probes in water. J. Chem. Theory Comput. 2021, 17, 6366–6386. [Google Scholar] [CrossRef]
- Akter, M.; Drinkwater, N.; Devine, S.M.; Drew, S.C.; Krishnarjuna, B.; Debono, C.O.; Wang, G.; Scanlon, M.J.; Scammels, P.J.; McGowan, S.; et al. Identification of the Binding Site of Apical Membrane Antigen 1 (AMA1) Inhibitors Using a Paramagnetic Probe. ChemMedChem 2019, 14, 603–612. [Google Scholar] [CrossRef] [Green Version]
- Kossman, S.; Kirchner, B.; Neese, F. Performance of modern density functional theory for the prediction of hyperfine structure: meta-GGA and double hybrid functionals. Mol. Phys. 2007, 105, 2049–2071. [Google Scholar] [CrossRef] [Green Version]
- Puzzarini, C.; Barone, V. Toward spectroscopic accuracy for organic free radicals: Molecular structure, vibrational spectrum and magnetic properties of F2NO. J. Chem. Phys. 2008, 129, 084306. [Google Scholar] [CrossRef] [PubMed]
- Houriez, C.; Ferré, N.; Siri, D.; Masella, M. Further insights into the environmental effects on the computed hyperfine coupling constants of nitroxides in aqueous solution. J. Phys. Chem. B 2009, 113, 15047–15056. [Google Scholar] [CrossRef]
- Houriez, C.; Masella, M.; Ferré, N. Structural and atoms-in-molecules analysis of hydrogen-bond network around nitroxides in liquid water. J. Chem. Phys. 2010, 133, 124508. [Google Scholar] [CrossRef] [PubMed]
- Puzzarini, C.; Bloino, J.; Tasinato, N.; Barone, V. Accuracy and Interpretability: The Devil and the Holy Grail. New Routes Across Old Boundaries in Computational Spectroscopy. Chem. Rev. 2019, 119, 8131–8191. [Google Scholar] [CrossRef]
- Biczysko, M.; Panek, P.; Scalmani, G.; Bloino, J.; Barone, V. Harmonic and anharmonic vibrational frequency calculations with the double-Hybrid B2PLYP method: Analytic second derivatives and benchmark studies. J. Chem. Theory Comput. 2010, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Ceselin, G.; Barone, V.; Tasinato, N. Accurate biomolecular structures by the nano-LEGO approach: Pick the bricks and build your geometry. J. Chem. Theory Comput. 2021, 17, 7290–7311. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. iii. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. Phys. Chem. Chem. Phys. 2014, 16, 1759–1787. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, 034108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papajak, E.; Leverentz, H.R.; Zheng, J.; Truhlar, D.G. Efficient diffuse basis sets: cc-pVxZ+ and maug-cc-pVxZ. J. Chem. Theory Comput. 2009, 5, 1197–1202. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Light, J.C.; Hamilton, I.P.; Lill, J.V. Generalized discrete variable approximation in quantum mechanics. J. Chem. Phys. 1985, 82, 1400–1409. [Google Scholar] [CrossRef]
- Colbert, D.T.; Miller, W.H. A novel discrete variable representation for quantum mechanical reactive scattering via the S-matrix Kohn method. J. Chem. Phys. 1992, 96, 1982–1991. [Google Scholar] [CrossRef] [Green Version]
- Bačić, Z.; Light, J.C. Theoretical methods for rovibrational states of floppy molecules. Ann. Rev. Phys. Chem. 1989, 40, 469–498. [Google Scholar] [CrossRef]
- Light, J.C.; Carrington, T., Jr. Discrete-variable representations and their utilization. In Advances in Chemical Physics; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2000; Volume 114, Chapter 4; pp. 263–310. [Google Scholar] [CrossRef]
- Baiardi, A.; Bloino, J.; Barone, V. Simulation of vibronic spectra of flexible systems: Hybrid DVR-harmonic approaches. J. Chem. Theory Comput. 2017, 13, 2804–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spackman, P.R.; Jayatilaka, D.; Karton, A. Basis set convergence of CCSD(T) equilibrium geometries using a large and diverse set of molecular structures. J. Chem. Phys. 2016, 145, 104101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Heßelmann, A.; Kats, D.; Köhn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [Green Version]
- Stanton, J.F.; Gauss, J.; Harding, M.E.; Szalay, P.G.; CFOUR. A Quantum Chemical Program Package. 2016. Available online: http://www.cfour.de (accessed on 2 December 2021).
- Penocchio, E.; Piccardo, M.; Barone, V. Semiexperimental equilibrium structures for building blocks of organic and biological molecules: The B2PLYP route. J. Chem. Theory Comput. 2015, 11, 4689–4707. [Google Scholar] [CrossRef] [PubMed]
- Knizia, G.; Adler, T.B.; Werner, H.J. Simplified CCSD(T)-F12 methods: Theory and benchmarks. J. Chem. Phys. 2009, 130, 054104. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Adler, T.B.; Werner, H.J. Systematically convergent basis sets for explicitly correlated wavefunctions: The atoms H, He, B–Ne, and Al–Ar. J. Chem. Phys. 2008, 128, 084102. [Google Scholar] [CrossRef]
- Alonso, E.R.; Fusè, M.; León, I.; Puzzarini, C.; Alonso, J.L.; Barone, V. Exploring the maze of cycloserine conformers in the gas phase guided by microwave spectroscopy and quantum chemistry. J. Phys. Chem. A 2021, 125, 2121–2129. [Google Scholar] [CrossRef]
- Briere, R.; Claxton, T.A.; Ellinger, Y.; Rey, P.; Laugier, J. Orientation of hyperfine tensors with respect to chemical bonds. Experimental and ab initio SCF + CI study in the nitroxide series. J. Am. Chem. Soc. 1982, 104, 34–38. [Google Scholar] [CrossRef]
- Komaromi, I.; Tronchet, J.M.J. Factors affecting the geometry (pyramidal vs. planar) of aminoxyl radicals. An ab initio study. J. Phys. Chem. 1995, 99, 10213–10220. [Google Scholar] [CrossRef]
- Cremer, D.; Pople, J.A. General definition of ring puckering coordinates. J. Am. Chem. Soc. 1975, 97, 1354–1358. [Google Scholar] [CrossRef]
- Rintoul, L.; Micallef, A.; Bottle, S. The vibrational group frequency of the N–O stretching band of nitroxide stable free radicals. Spectrochim. Acta A 2008, 70, 713–717. [Google Scholar] [CrossRef] [Green Version]
- Vreven, T.; Morokuma, K. Chapter 3 Hybrid Methods: ONIOM(QM:MM) and QM/MM. Ann. Rep. Comput. Chem. 2006, 2, 35–51. [Google Scholar]
- Yang, Q.; Mendolicchio, M.; Barone, V.; Bloino, J. Accuracy and reliability in the simulation of vibrational spectra: A comprehensive benchmark of energies and intensities issuing from generalized vibrational perturbation theory to second order (GVPT2). Front. Astron. Space Sci. 2021, 8, 665232. [Google Scholar] [CrossRef]
- Stipa, P. A multi-step procedure for evaluating the EPR parameters of indolinonic aromatic aminoxyls: A combined DFT and spectroscopic study. Chem. Phys. 2006, 323, 501–510. [Google Scholar]
- Del Galdo, S.; Fusè, M.; Barone, V. The ONIOM/PMM model for effective yet accurate simulation of optical and chiroptical spectra in solution: Camphorquinone in methanol as a case study. J. Chem. Theory Comput. 2020, 16, 3294–3306. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
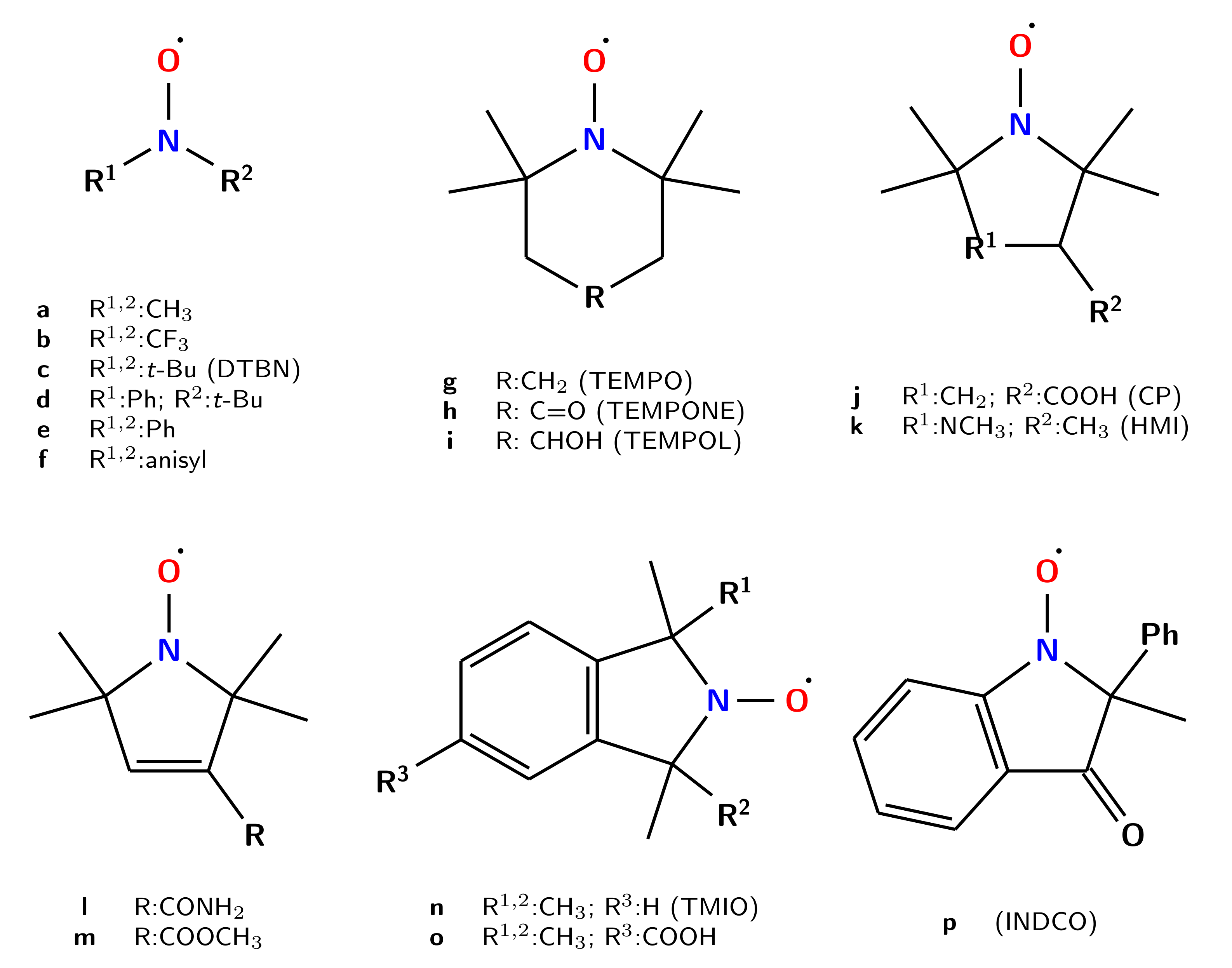{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Minimum | Transition State | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Level of Theory | Basis Sets | rNO | rCN | CNC | CCNO | rNO | rCN | CNC | CCNO |
| B3 | SNSD | 1.2798 | 1.4593 | 118.6 | 153.9 | 1.2804 | 1.4545 | 119.4 | 180.0 |
| B2 | maugTZ | 1.2796 | 1.4549 | 118.7 | 153.3 | 1.2802 | 1.4504 | 119.3 | 180.0 |
| CCSD(T)-F12 | DZ-F12 | 1.2773 | 1.4554 | 118.3 | 150.8 | 1.2774 | 1.4491 | 119.6 | 180.0 |
| MeNO | |||
|---|---|---|---|
| Level of Theory | Basis Sets | Minimum | TS |
| B3 | EPR(III) | 39.38 | 24.46 |
| BHLYP | EPR(III) | 43.63 | 31.54 |
| B2 | EPR(III) | 44.21 | 29.26 |
| CCSD(T) | EPR(III) | 44.26 | 30.20 |
| Mol | NO | CN | CN | CNC | CNO | CNO | CCNO |
|---|---|---|---|---|---|---|---|
| a | 1.280 | 1.455 | 118.70 | 153.30 | |||
| b | 1.267 | 1.460 | 1.470 | 120.98 | 117.37 | 120.18 | 165.77 |
| c | 1.284 | 1.507 | 1.506 | 127.26 | 116.18 | 113.10 | 158.02 |
| d | 1.282 | 1.503 | 1.420 | 125.17 | 117.79 | 117.01 | 177.76 |
| e | 1.283 | 1.421 | 123.13 | 118.43 | 180.00 | ||
| f | 1.286 | 1.418 | 123.06 | 118.47 | 180.00 | ||
| g | 1.282 | 1.495 | 124.11 | 115.81 | 155.75 | ||
| h | 1.280 | 1.496 | 124.06 | 115.62 | 154.56 | ||
| i | 1.281 | 1.494 | 123.97 | 115.87 | 155.74 | ||
| j | 1.271 | 1.481 | 1.483 | 115.55 | 121.88 | 122.50 | 176.97 |
| k | 1.271 | 1.476 | 1.475 | 113.90 | 122.43 | 123.39 | 174.11 |
| l | 1.270 | 1.487 | 1.481 | 114.47 | 122.54 | 122.14 | 169.69 |
| m | 1.270 | 1.486 | 1.480 | 114.84 | 122.71 | 122.45 | 180.00 |
| n | 1.270 | 1.485 | 1.485 | 115.63 | 122.22 | 122.15 | 180.000 |
| o | 1.271 | 1.485 | 1.485 | 115.59 | 122.21 | 122.21 | 180.00 |
| p | 1.270 | 1.484 | 1.389 | 111.71 | 123.68 | 124.57 | 177.83 |
| Mol. | ||||||
|---|---|---|---|---|---|---|
| b | 1397 | 1439 | 1410 | 29 | 1437 | 1408 |
| c | 1342 | 1384 | 1350 | 34 | 1359 | 1325 |
| d | 1370 | 1412 | 1379 | 33 | 1387 | 1354 |
| e | 1342 | 1392 | 1360 | 32 | 1379 | 1347 |
| f | 1346 | 1400 | 1369 | 31 | 1392 | 1361 |
| g | 1339 | 1377 | 1345 | 31 | 1368 | 1336 |
| h | 1380 | 1392 | 1362 | 30 | 1398 | 1367 |
| i | 1371 | 1401 | 1370 | 31 | 1397 | 1366 |
| j | - | 1472 | 1440 | 32 | 1453 | 1421 |
| k | - | 1485 | 1456 | 29 | 1461 | 1432 |
| l | 1438 e | 1481 | 1453 | 28 | 1457 | 1429 |
| m | 1435 e | 1487 | 1455 | 32 | 1460 | 1427 |
| n | 1428 e | 1486 | 1452 | 34 | 1458 | 1424 |
| o | 1427 | 1488 | 1454 | 34 | 1465 | 1431 |
| p | - | 1468 | 1438 | 30 | 1456 | 1426 |
| Mol | Modes | ||
|---|---|---|---|
| b | 29 | 29 | 7 |
| d | 33 | 35 | 11 |
| e | 32 | 33 | 11 |
| f | 31 | 32 | 7 |
| h | 30 | 30 | 8 |
| i | 30 | 35 | 7 |
| Mol | NO | a | TM | ||||
|---|---|---|---|---|---|---|---|
| b | 1.267 | 1.4 | 12.26 | 23.12 | 2.71 | 0.68 | 0.63 |
| c | 1.284 | 8.0 | 20.13 | 40.14 | 1.66 | 0.44 | 0.10 |
| d | 1.282 | 5.6 | 2.00 | 25.54 | 2.62 | 1.05 | 0.94 |
| e | 1.283 | 3.6 | 0.00 | 20.11 | 1.10 | 0.99 | 0.95 |
| f | 1.286 | 3.5 | 0.00 | 21.10 | 2.73 | 0.99 | 0.95 |
| g | 1.282 | 5.1 | 21.70 | 40.42 | 1.14 | 0.22 | −0.03 |
| h | 1.280 | 5.2 | 22.79 | 40.62 | 2.07 | 0.09 | −0.12 |
| i | 1.281 | 4.9 | 21.70 | 40.18 | 1.34 | 0.20 | −0.03 |
| j | 1.271 | −4.0 | 2.60 | 30.82 | 6.23 | 1.03 | 0.93 |
| k | 1.271 | −5.7 | 4.97 | 31.21 | 7.17 | 0.99 | 0.89 |
| l | 1.270 | −5.1 | 8.68 | 32.83 | 6.22 | 0.82 | 0.78 |
| m | 1.270 | −4.7 | 0.00 | 30.66 | 5.21 | 0.98 | 0.95 |
| n | 1.271 | −3.9 | 0.00 | 30.64 | 2.70 | 0.98 | 0.95 |
| o | 1.270 | −3.9 | 0.00 | 30.45 | 5.47 | 0.99 | 0.95 |
| p | 1.270 | −7.8 | 1.80 | 20.27 | 4.40 | 0.98 | 0.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barone, V.; Fusè, M.; Pinto, S.M.V.; Tasinato, N. A Computational Journey across Nitroxide Radicals: From Structure to Spectroscopic Properties and Beyond. Molecules 2021, 26, 7404. https://doi.org/10.3390/molecules26237404
Barone V, Fusè M, Pinto SMV, Tasinato N. A Computational Journey across Nitroxide Radicals: From Structure to Spectroscopic Properties and Beyond. Molecules. 2021; 26(23):7404. https://doi.org/10.3390/molecules26237404
Chicago/Turabian StyleBarone, Vincenzo, Marco Fusè, Sandra Mónica Vieira Pinto, and Nicola Tasinato. 2021. "A Computational Journey across Nitroxide Radicals: From Structure to Spectroscopic Properties and Beyond" Molecules 26, no. 23: 7404. https://doi.org/10.3390/molecules26237404
APA StyleBarone, V., Fusè, M., Pinto, S. M. V., & Tasinato, N. (2021). A Computational Journey across Nitroxide Radicals: From Structure to Spectroscopic Properties and Beyond. Molecules, 26(23), 7404. https://doi.org/10.3390/molecules26237404