
Ab Initio Study of Fine and Hyperfine Interactions in Triplet POH

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
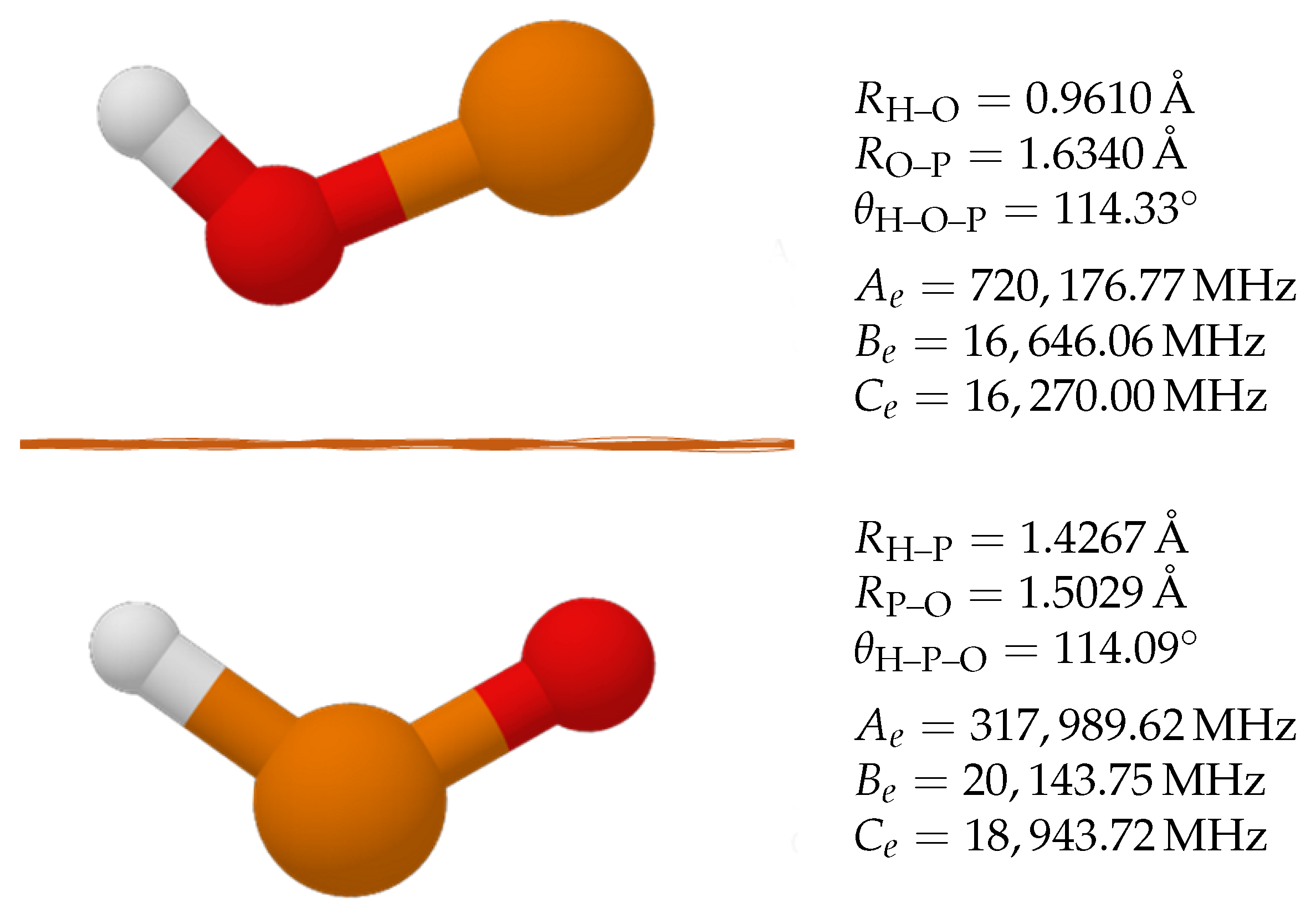
2.1. Molecular Structure
2.2. Spectroscopic Parameters
3. Results
3.1. Molecular Structure
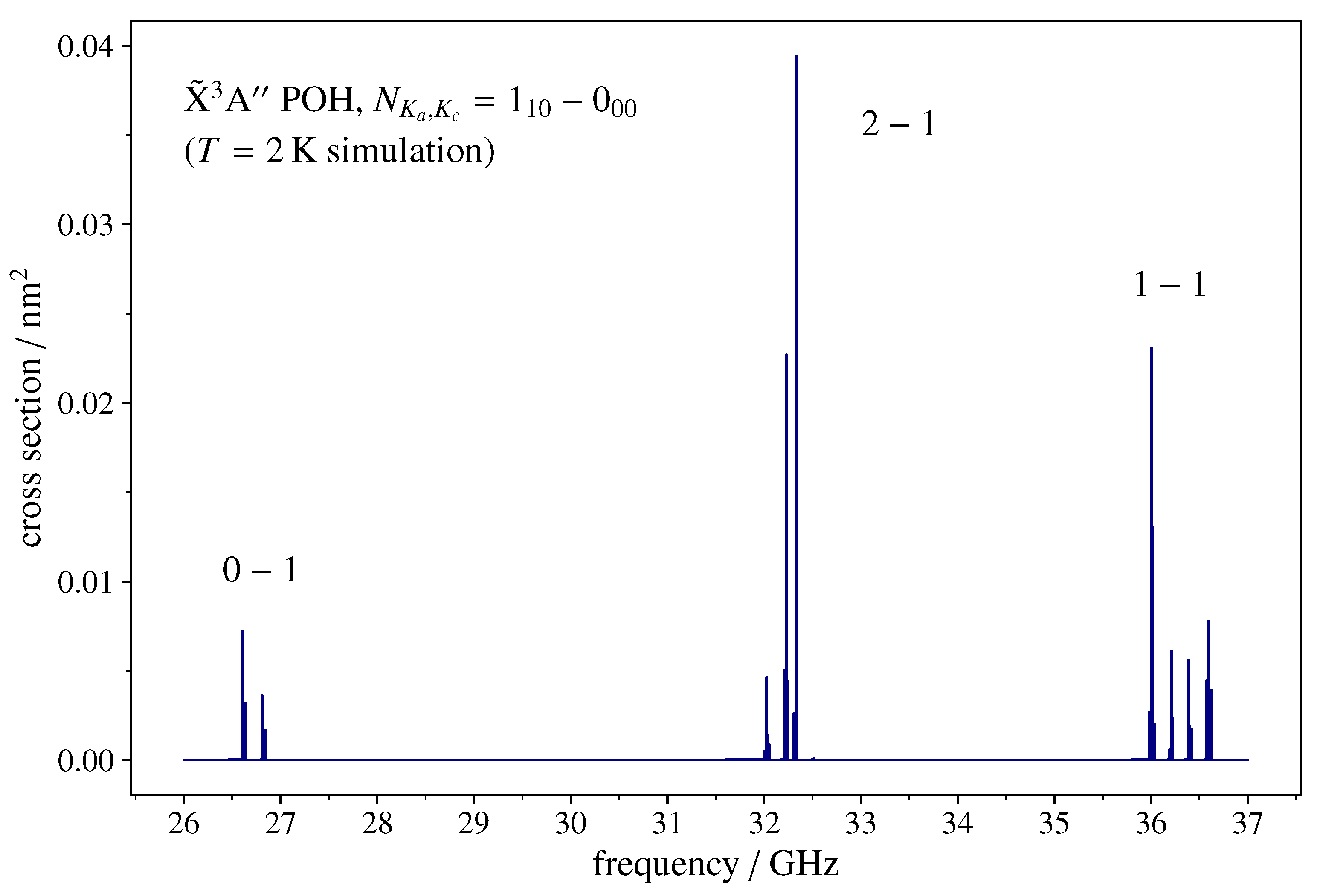
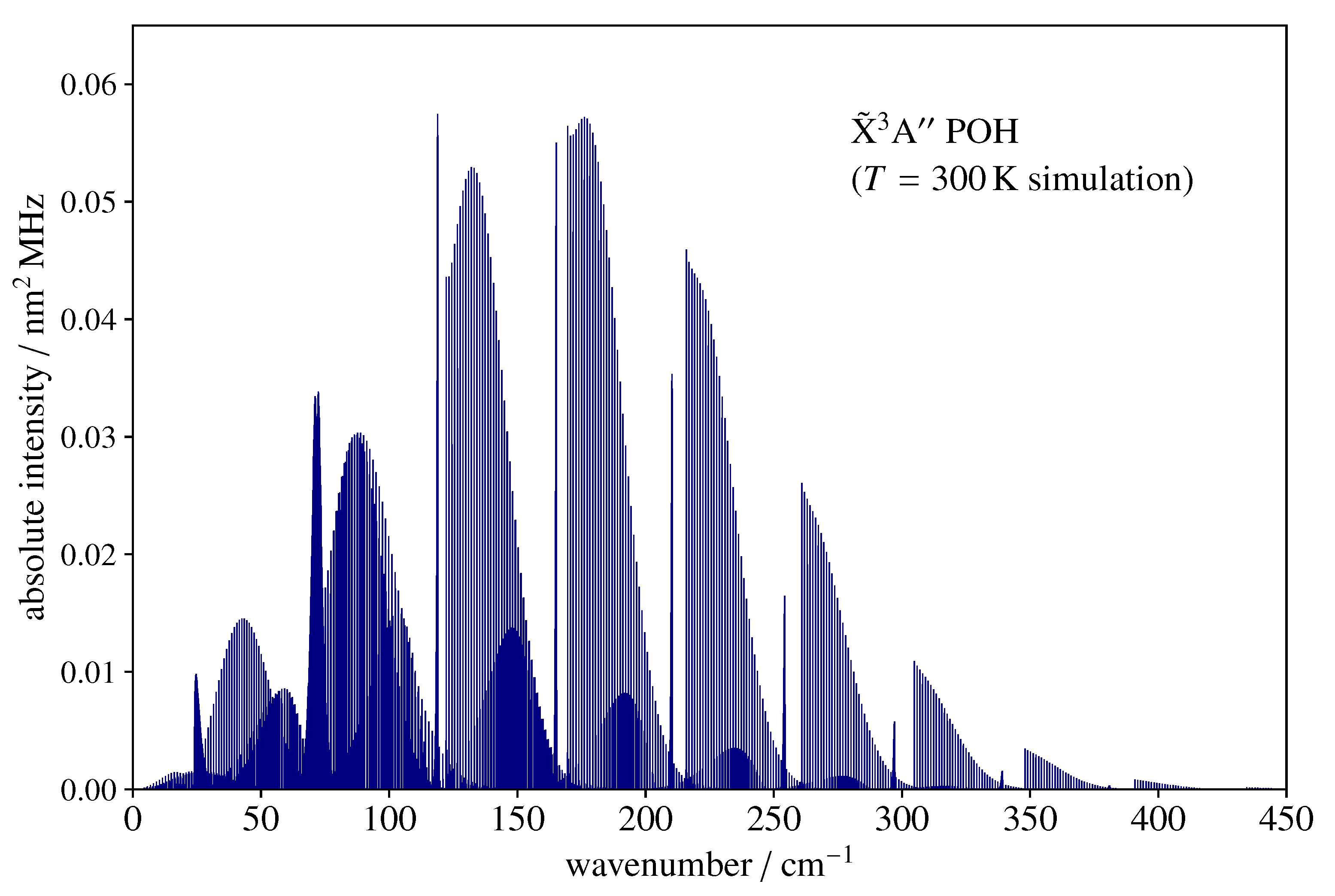
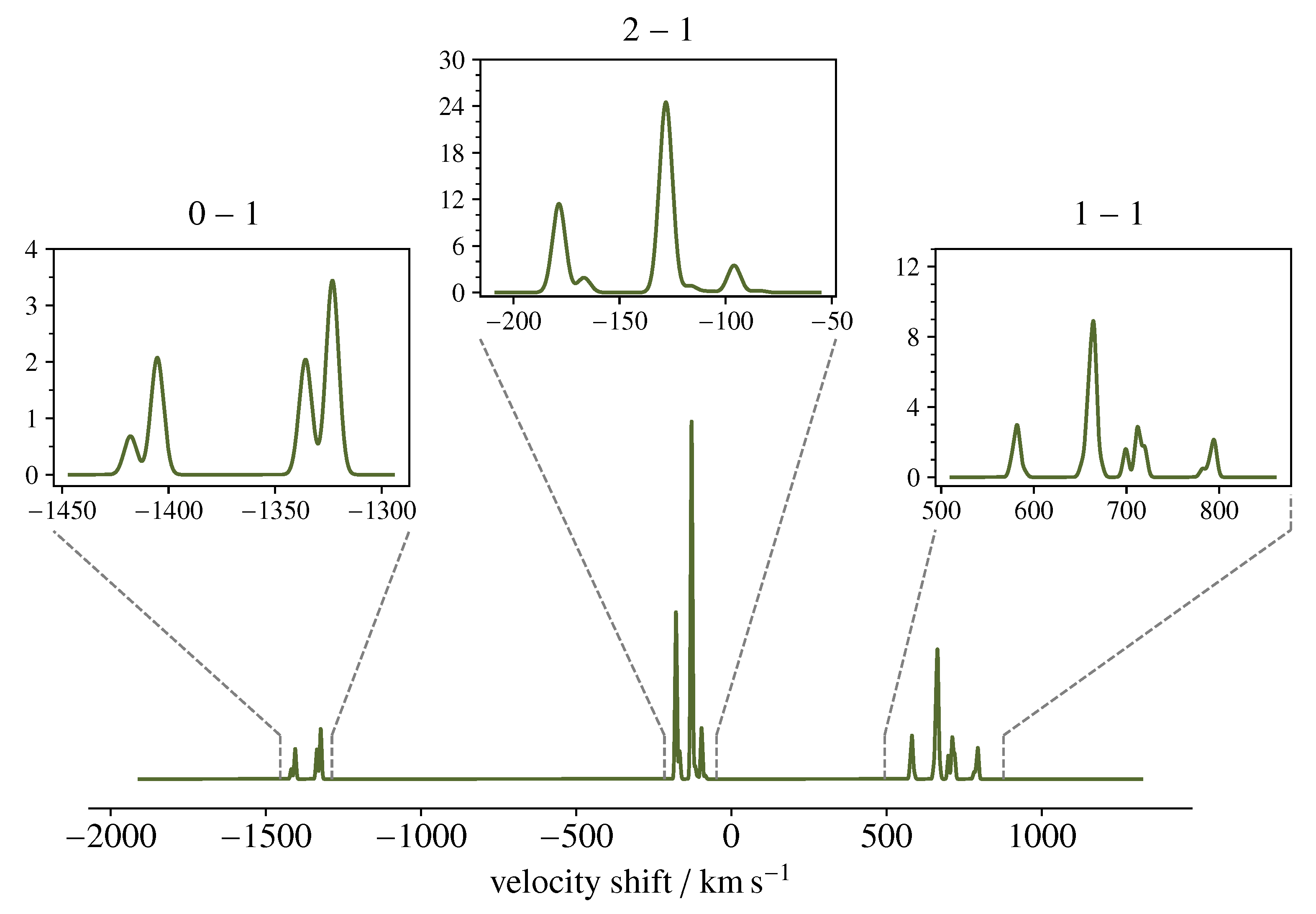
3.2. Spectral Simulations
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Maciá, E. The Chemical Evolution of Phosphorus an Interdisciplinary Approach to Astrobiology; Apple Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- Powner, M.W.; Gerland, B.; Sutherland, J.D. Synthesis of activated pyrimidine ribonucleotides in prebiotically plausible conditions. Nature 2009, 459, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Chyba, C.F.; Thomas, P.J.; Brookshaw, L.; Sagan, C. Cometary Delivery of Organic Molecules to the Early Earth. Science 1990, 249, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.A. Rethinking early Earth phosphorus geochemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 853–858. [Google Scholar] [CrossRef] [Green Version]
- Rivilla, V.M.; Drozdovskaya, M.N.; Altwegg, K.; Caselli, P.; Beltrán, M.T.; Fontani, F.; van der Tak, F.F.S.; Cesaroni, R.; Vasyunin, A.; Rubin, M.; et al. ALMA and ROSINA detections of phosphorus-bearing molecules: The interstellar thread between star-forming regions and comets. Mon. Not. R. Astron. Soc. 2020, 492, 1180–1198. [Google Scholar] [CrossRef]
- Turner, B.E.; Bally, J. Detection of Interstellar PN: The First Identified Phosphorus Compound in the Interstellar Medium. Astrophys. J. Lett. 1987, 321, L75. [Google Scholar] [CrossRef]
- Ziurys, L.M. Detection of Interstellar PN: The First Phosphorus-bearing Species Observed in Molecular Clouds. Astrophys. J. Lett. 1987, 321, L81. [Google Scholar] [CrossRef]
- Rivilla, V.; Fontani, F.; Beltrán, M.; Vasyunin, A.; Caselli, P.; Martín-Pintado, J.; Cesaroni, R. The first detections of the key prebiotic molecule PO in star-forming regions. Astrophys. J. 2016, 826, 161. [Google Scholar] [CrossRef]
- Lefloch, B.; Vastel, C.; Viti, S.; Jimenez-Serra, I.; Codella, C.; Podio, L.; Ceccarelli, C.; Mendoza, E.; Lepine, J.R.D.; Bachiller, R. Phosphorus-bearing molecules in solar-type star-forming regions: First PO detection. Mon. Not. R. Astron. Soc. 2016, 462, 3937–3944. [Google Scholar] [CrossRef] [Green Version]
- Bergner, J.B.; Öberg, K.I.; Walker, S.; Guzmán, V.V.; Rice, T.S.; Bergin, E.A. Detection of Phosphorus-bearing Molecules toward a Solar-type Protostar. Astrophys. J. Lett. 2019, 884, L36. [Google Scholar] [CrossRef]
- Rivilla, V.M.; Jiménez-Serra, I.; Zeng, S.; Martín, S.; Martín-Pintado, J.; Armijos-Abendaño, J.; Viti, S.; Aladro, R.; Riquelme, D.; Requena-Torres, M.; et al. Phosphorus-bearing molecules in the Galactic Center. Mon. Not. R. Astron. Soc. 2018, 475, L30–L34. [Google Scholar] [CrossRef]
- Fontani, F.; Rivilla, V.M.; van der Tak, F.F.S.; Mininni, C.; Beltrán, M.T.; Caselli, P. Origin of the PN molecule in star-forming regions: The enlarged sample. Mon. Not. R. Astron. Soc. 2019, 489, 4530–4542. [Google Scholar] [CrossRef]
- McGuire, B.A. 2018 census of interstellar, circumstellar, extragalactic, protoplanetary disk, and exoplanetary molecules. Astrophys. J. Suppl. Ser. 2018, 239, 17. [Google Scholar] [CrossRef]
- Puzzarini, C.; Stanton, J.F.; Gauss, J. Quantum-chemical calculation of spectroscopic parameters for rotational spectroscopy. Int. Rev. Phys. Chem. 2010, 29, 273–367. [Google Scholar] [CrossRef]
- Puzzarini, C. Rotational spectroscopy meets theory. Phys. Chem. Chem. Phys. 2013, 15, 6595–6607. [Google Scholar] [CrossRef]
- Puzzarini, C.; Bloino, J.; Tasinato, N.; Barone, V. Accuracy and interpretability: The devil and the holy grail. New routes across old boundaries in computational spectroscopy. Chem. Rev. 2019, 119, 8131–8191. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Alessandrini, S.; Biczysko, M.; Cheeseman, J.R.; Clary, D.C.; McCoy, A.B.; DiRisio, R.J.; Neese, F.; Melosso, M.; Puzzarini, C. Computational molecular spectroscopy. Nat. Rev. Methods Prim. 2021, 1, 1–27. [Google Scholar] [CrossRef]
- Endres, C.P.; Schlemmer, S.; Schilke, P.; Stutzki, J.; Müller, H.S.P. The Cologne Database for Molecular Spectroscopy, CDMS, in the Virtual Atomic and Molecular Data Centre, VAMDC. J. Mol. Spectrosc. 2016, 327, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Gordy, W.; Cook, R.L. Microwave Molecular Spectra; Wiley: New York, NY, USA, 1984. [Google Scholar]
- Kroto, H.W. Molecular Rotation Spectra; Wiley: London, UK, 1975. [Google Scholar]
- Puzzarini, C.; Barone, V. Diving for Accurate Structures in the Ocean of Molecular Systems with the Help of Spectroscopy and Quantum Chemistry. Acc. Chem. Res. 2018, 51, 548–556. [Google Scholar] [CrossRef] [Green Version]
- Peña, I.; Sanz, M.E.; López, J.C.; Alonso, J.L. Preferred Conformers of Proteinogenic Glutamic Acid. J. Am. Chem. Soc. 2012, 134, 2305–2312. [Google Scholar] [CrossRef]
- Cazzoli, G.; Puzzarini, C. The Lamb-dip spectrum of the J+1←J (J = 0, 1, 3–8) transitions of H13CN: The nuclear hyperfine structure due to H, 13C, and 14N. J. Mol. Spectrosc. 2005, 233, 280–289. [Google Scholar] [CrossRef]
- Cazzoli, G.; Puzzarini, C. Sub-Doppler Resolution in the THz Frequency Domain: 1 kHz Accuracy at 1 THz by Exploiting the Lamb-Dip Technique. J. Phys. Chem. A 2013, 117, 13759–13766. [Google Scholar] [CrossRef] [PubMed]
- Puzzarini, C.; Cazzoli, G.; Harding, M.E.; Vázquez, J.; Gauss, J. A new experimental absolute nuclear magnetic shielding scale for oxygen based on the rotational hyperfine structure of H217O. J. Chem. Phys. 2009, 131, 234304. [Google Scholar] [CrossRef]
- Melosso, M.; Melli, A.; Spada, L.; Zheng, Y.; Chen, J.; Li, M.; Lu, T.; Feng, G.; Gou, Q.; Dore, L.; et al. Rich Collection of n-Propylamine and Isopropylamine Conformers: Rotational Fingerprints and State-of-the-Art Quantum Chemical Investigation. J. Phys. Chem. A 2020, 124, 1372–1381. [Google Scholar] [CrossRef]
- Francisco, J.S. Accurate ab initio spectroscopic properties of HOPx and HPOx (x = −1, 0, +1). Chem. Phys. 2003, 287, 303–316. [Google Scholar] [CrossRef]
- Puzzarini, C. Theoretical study of XPO (X = H, F, Cl, Br) molecules: Structural and molecular properties. J. Mol. Struct. 2006, 780–781, 238–246. [Google Scholar] [CrossRef]
- Saito, S.; Endo, Y.; Hirota, E. The microwave spectrum of an unstable molecule, HPO. J. Chem. Phys. 1986, 84, 1157–1159. [Google Scholar] [CrossRef]
- García de la Concepción, J.; Puzzarini, C.; Barone, V.; Jiménez-Serra, I.; Roncero, O. Formation of phosphorus monoxide (PO) in the interstellar medium: Insights from quantum-chemical and kinetic calculations. Astrophys. J. 2021, 922, 169. [Google Scholar] [CrossRef]
- Cook, R.L.; De Lucia, F.C. Application of the Theory of Irreducible Tensor Operators to Molecular Hyperfine Structure. Am. J. Phys. 1971, 39, 1433–1454. [Google Scholar] [CrossRef]
- Bowater, I.C.; Brown, J.M.; Carrington, A. Microwave Spectroscopy of Nonlinear Free Radicals. I. General Theory and Application to the Zeeman Effect in HCO. Proc. R. Soc. Lond. 1973, 333, 265–288. [Google Scholar]
- Heckert, M.; Kállay, M.; Gauss, J. Molecular Equilibrium Geometries Based on Coupled-Cluster Calculations Including Quadruple Excitations. Mol. Phys. 2005, 103, 2109. [Google Scholar] [CrossRef]
- Heckert, M.; Kállay, M.; Tew, D.P.; Klopper, W.; Gauss, J. Basis-Set Extrapolation Techniques for the Accurate Calculation of Molecular Equilibrium Geometries Using Coupled-Cluster Theory. J. Chem. Phys. 2006, 125, 044108. [Google Scholar] [CrossRef] [PubMed]
- Stanton, J.F.; Gauss, J.; Harding, M.E.; Szalay, P.G. CFOUR, Coupled-Cluster Techiques for Computational Chemistry. 2008. Available online: http://cfour.de (accessed on 1 December 2021).
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Feller, D. The use of systematic sequences of wave functions for estimating the complete basis set, full configuration interaction limit in water. J. Chem. Phys. 1993, 98, 7059–7071. [Google Scholar] [CrossRef]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639–9646. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Wilson, A.K.; van Mourik, T.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J. Mol. Struct. THEOCHEM 1996, 388, 339–349. [Google Scholar] [CrossRef]
- Van Mourik, T.; Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. VIII. Standard and augmented sextuple zeta correlation consistent basis sets for aluminum through argon. Int. J. Quantum Chem. 2000, 76, 205–221. [Google Scholar] [CrossRef]
- Peterson, K.A.; Dunning, T.H., Jr. Accurate correlation consistent basis sets for molecular core-valence correlation effects: The second row atoms Al-Ar, and the first row atoms B-Ne revisited. J. Chem. Phys. 2002, 117, 10548–10560. [Google Scholar] [CrossRef] [Green Version]
- Noga, J.; Bartlett, R.J. The Full CCSDT Model for Molecular Electronic Structure. J. Chem. Phys. 1987, 86, 7041–7050. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Schaefer, H.F., III. A New Implementation of the Full CCSDT Model for Molecular Electronic-Structure. Chem. Phys. Lett. 1988, 152, 382–386. [Google Scholar] [CrossRef]
- Watts, J.D.; Bartlett, R.J. The Coupled-Cluster Single, Double, and Triple Excitation Model for Open-Shell Single Reference Functions. J. Chem. Phys. 1990, 93, 6104–6105. [Google Scholar] [CrossRef]
- Kállay, M.; Surján, P.R. Higher Excitations in Coupled-Cluster Theory. J. Chem. Phys. 2001, 115, 2945–2954. [Google Scholar] [CrossRef]
- Kállay, M.; Nagy, P.R.; Rolik, Z.; Mester, D.; Samu, G.; Csontos, J.; Csóka, J.; Szabó, B.P.; Gyevi-Nagy, L.; Ladjánszki, I.; et al. MRCC, a Quantum Chemical Program Suite. 2018. Available online: http://www.mrcc.hu (accessed on 11 November 2021).
- Mills, I.M. Vibration-Rotation Structure in Asymmetric- and Symmetric-Top Molecules. In Molecular Spectroscopy: Modern Research; Rao, K.N., Matthews, C.W., Eds.; Academic Press: Cambridge, MA, USA, 1972; Volume 1, pp. 115–140. [Google Scholar]
- Vahtras, O.; Loboda, O.; Minaev, B.; Agren, H.; Ruud, K. Ab initio calculations of zero-field splitting parameters. Chem. Phys. 2002, 279, 133–142. [Google Scholar] [CrossRef]
- Aidas, K.; Angeli, C.; Bak, K.L.; Bakken, V.; Bast, R.; Boman, L.; Christiansen, O.; Cimiraglia, R.; Coriani, S.; Dahle, P.; et al. The Dalton quantum chemistry program system. WIREs Comput. Mol. Sci. 2014, 4, 269–284. [Google Scholar] [CrossRef]
- Dalton, a Molecular Electronic Structure Program, Release v2020.0.1 (2021). Available online: http://daltonprogram.org (accessed on 11 November 2021).
- Van Vleck, J.H. The Coupling of Angular Momentum Vectors in Molecules. Rev. Mod. Phys. 1951, 23, 213–227. [Google Scholar] [CrossRef]
- Fermi, E. Über die magnetischen Momente der Atomkerne. Z. Phys. 1930, 60, 320–333. [Google Scholar] [CrossRef]
- Frosch, R.A.; Foley, H.M. Magnetic Hyperfine Structure in Diatomic Molecules. Phys. Rev. 1952, 88, 1337–1349. [Google Scholar] [CrossRef]
- Tarczay, G.; Szalay, P.G.; Gauss, J. First-Principles Calculation of Electron Spin-Rotation Tensors. J. Phys. Chem. A 2010, 114, 9246–9252. [Google Scholar] [CrossRef] [PubMed]
- Gauss, J.; Ruud, K.; Helgaker, T. Perturbation-dependent atomic orbitals for the calculation of spin-rotation constants and rotational g tensors. J. Chem. Phys. 1996, 105, 2804–2812. [Google Scholar] [CrossRef]
- Gauss, J.; Stanton, J.F. Perturbative treatment of triple excitations in coupled-cluster calculations of nuclear magnetic shielding constants. J. Chem. Phys. 1996, 104, 2574–2583. [Google Scholar] [CrossRef]
- Gauss, J.; Sundholm, D. Coupled-cluster calculations of spin-rotation constants. Mol. Phys. 1997, 91, 449–458. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.K.G. Vibrational Spectra and Structure; Durig, J., Ed.; Elsevier: Amsterdam, The Netherlands, 1977; Volume 6, pp. 1–89. [Google Scholar]
- Brown, J.M.; Carrington, A. Rotational Spectroscopy of Diatomic Molecules; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Sharma, K.; Miller, T.A.; Stanton, J.F. Vibronically coupled states: Computational considerations and characterisation of vibronic and rovibronic spectroscopic parameters. Int. Rev. Phys. Chem. 2021, 40, 165–298. [Google Scholar] [CrossRef]
- Melosso, M.; Bizzocchi, L.; Sipilä, O.; Giuliano, B.; Dore, L.; Tamassia, F.; Martin-Drumel, M.A.; Pirali, O.; Redaelli, E.; Caselli, P. First detection of NHD and ND2 in the interstellar medium-Amidogen deuteration in IRAS 16293–2422. Astron. Astrophys. 2020, 641, A153. [Google Scholar] [CrossRef]
- Alessandrini, S.; Gauss, J.; Puzzarini, C. Accuracy of rotational parameters predicted by high-level quantum-chemical calculations: Case study of sulfur-containing molecules of astrochemical interest. J. Chem. Theory Comput. 2018, 14, 5360–5371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melosso, M.; Diouf, M.L.; Bizzocchi, L.; Harding, M.E.; Cozijn, F.M.; Puzzarini, C.; Ubachs, W. Hyperfine-Resolved Near-Infrared Spectra of . J. Phys. Chem. A 2021, 125, 7884–7890. [Google Scholar] [CrossRef] [PubMed]
- Melosso, M.; Dore, L.; Gauss, J.; Puzzarini, C. Deuterium hyperfine splittings in the rotational spectrum of NH2D as revealed by Lamb-dip spectroscopy. J. Mol. Spectrosc. 2020, 370, 111291. [Google Scholar] [CrossRef]
- Puzzarini, C.; Barone, V. Toward spectroscopic accuracy for organic free radicals: Molecular structure, vibrational spectrum, and magnetic properties of F2NO. J. Chem. Phys. 2008, 129, 084306. [Google Scholar] [CrossRef]
- Puzzarini, C.; Barone, V. Toward spectroscopic accuracy for open-shell systems: Molecular structure and hyperfine coupling constants of H2CN, H2CP, NH2, and PH2 as test cases. J. Chem. Phys. 2010, 133, 184301. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HPO | HPO | Parameter | POH | POH |
|---|---|---|---|---|---|
| 1.4521 | 1.4244 | 0.9626 | 0.9605 | ||
| 1.4799 | 1.4989 | 1.6165 | 1.6346 | ||
| 104.54 | 114.88 | 111.79 | 113.92 |
| Parameter | Unit | HPO | POH |
|---|---|---|---|
| MHz | 311,681.77 | 734,899.81 | |
| MHz | 20,013.26 | 16,590.40 | |
| MHz | 18,694.47 | 16,184.70 | |
| MHz | 0.0247 | 0.0264 | |
| MHz | 2.57 | 2.20 | |
| MHz | 127.26 | 509.0 | |
| kHz | −1.47 | −0.53 | |
| kHz | −0.64 | −0.06 | |
| Hz | −0.12 | 0.012 | |
| kHz | 0.051 | 0.004 | |
| kHz | −4.65 | 5.87 | |
| kHz | 222.5 | 1666.20 | |
| mHz | 11.3 | 0.81 | |
| mHz | 9.24 | 0.93 | |
| mHz | 5.79 | 0.12 | |
| D | 2.45 | 0.65 | |
| D | 0.51 | 1.39 |
| Parameter | Unit | HPO | POH |
|---|---|---|---|
| D | MHz | 9043.22 | 9503.08 |
| E | MHz | 1538.29 | 11.23 |
| MHz | 604.16 | −41.22 | |
| MHz | −50.82 | 4.32 | |
| MHz | −116.53 | 10.01 | |
| MHz | −461.71 | −51.99 | |
| MHz | 759.78 | 115.90 | |
| MHz | −259.02 | −617.40 | |
| MHz | −116.71 | 334.04 | |
| MHz | 375.73 | 283.35 | |
| MHz | 183.90 | 10.05 | |
| kHz | 108.84 | 31.84 | |
| kHz | 49.80 | 45.46 | |
| kHz | 43.05 | 40.98 | |
| MHz | 196.92 | 24.64 | |
| MHz | −11.048 | 6.78 | |
| MHz | 9.712 | 6.73 | |
| MHz | 1.335 | −13.51 | |
| MHz | −17.741 | −20.36 | |
| kHz | −29.09 | −64.52 | |
| kHz | 2.03 | −0.27 | |
| kHz | 1.54 | −2.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bizzocchi, L.; Alessandrini, S.; Melosso, M.; Rivilla, V.M.; Puzzarini, C. Ab Initio Study of Fine and Hyperfine Interactions in Triplet POH. Molecules 2022, 27, 302. https://doi.org/10.3390/molecules27010302
Bizzocchi L, Alessandrini S, Melosso M, Rivilla VM, Puzzarini C. Ab Initio Study of Fine and Hyperfine Interactions in Triplet POH. Molecules. 2022; 27(1):302. https://doi.org/10.3390/molecules27010302
Chicago/Turabian StyleBizzocchi, Luca, Silvia Alessandrini, Mattia Melosso, Víctor M. Rivilla, and Cristina Puzzarini. 2022. "Ab Initio Study of Fine and Hyperfine Interactions in Triplet POH" Molecules 27, no. 1: 302. https://doi.org/10.3390/molecules27010302
APA StyleBizzocchi, L., Alessandrini, S., Melosso, M., Rivilla, V. M., & Puzzarini, C. (2022). Ab Initio Study of Fine and Hyperfine Interactions in Triplet POH. Molecules, 27(1), 302. https://doi.org/10.3390/molecules27010302