
Machine-Learning-Enabled Virtual Screening for Inhibitors of Lysine-Specific Histone Demethylase 1


 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Data Collection
2.2. Molecular Fingerprints
2.3. Model Construction
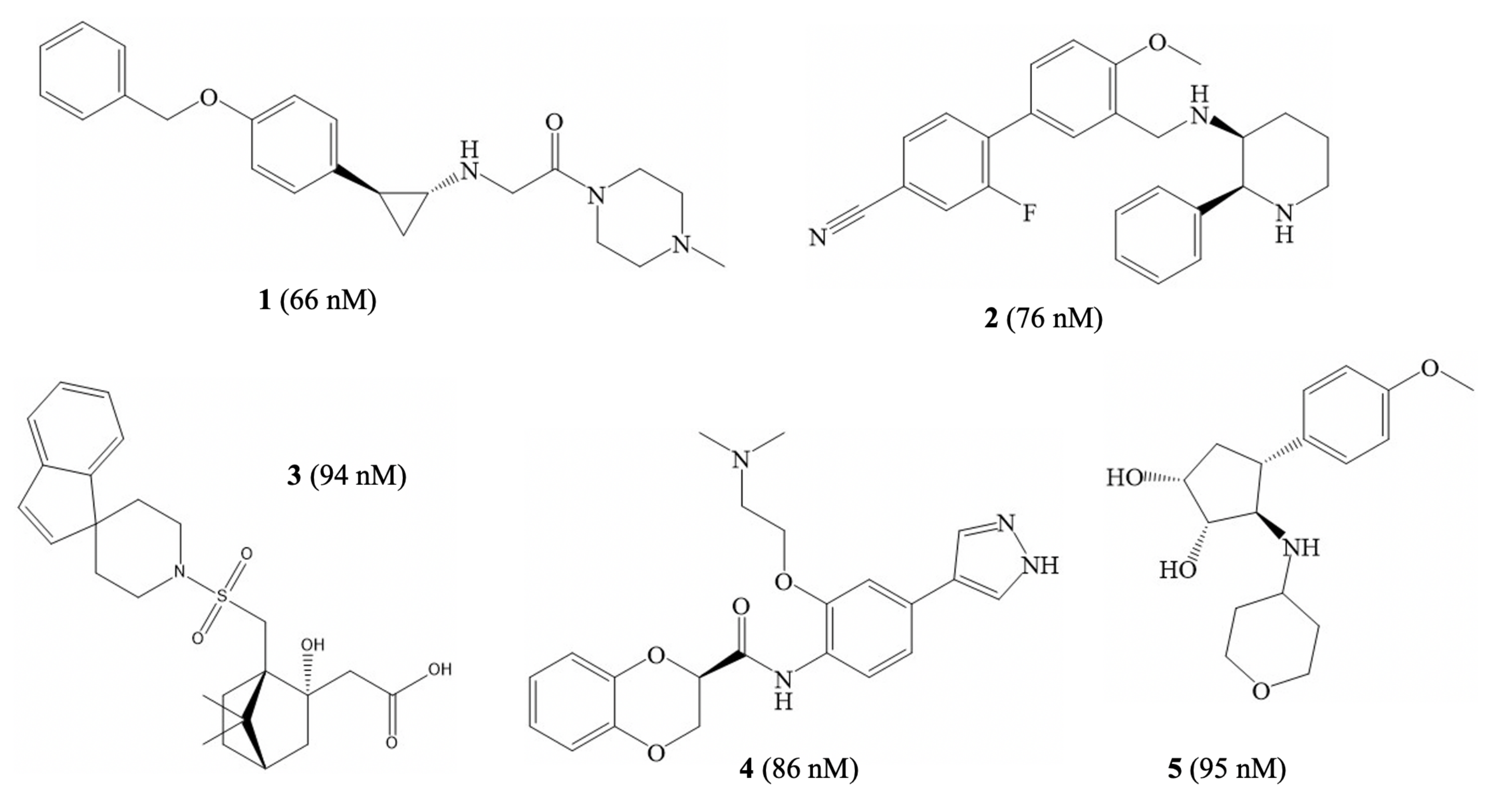
2.4. Virtual Screening
3. Results and Discussion
3.1. Characterisation of the Dataset
3.2. Performance of the Machine Learning Algorithms
3.3. Performance on Subsets of the Data
3.4. Virtual Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| DT | Decision tree regressor |
| HRP | Horseradish peroxidase |
| LSD1 | Lysine-specific histone demethylase 1 |
| MAO | Monoamine oxidases |
| MLP | Multi-layer perceptron |
| QSAR | Quantitative structure-activity relationship |
| RBF | Radial basis function |
| RF | Random forest regressor |
| RMSE | Root mean square error |
| SMILES | Simplified Molecular-Input Line-Entry System |
| SVR | Support vector regressor |
| t-SNE | t-distributed Stochastic Neighbour Embedding |
| TCP | Tranylcypromine |
References
- Wang, X.; Huang, B.; Suzuki, T.; Liu, X.; Zhan, P. Medicinal chemistry insights in the discovery of novel LSD1 inhibitors. Epigenomics 2015, 7, 1379–1396. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zhang, R.; Jiang, H.; Zhang, H.; Luo, C. Computer-aided drug design in epigenetics. Front. Chem. 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Sorna, V.; Theisen, E.R.; Stephens, B.; Warner, S.L.; Bearss, D.J.; Vankayalapati, H.; Sharma, S. High-throughput virtual screening identifies novel N′-(1-phenylethylidene)-benzohydrazides as potent, specific, and reversible LSD1 inhibitors. J. Med. Chem. 2013, 56, 9496–9508. [Google Scholar] [CrossRef]
- Gale, M.; Yan, Q. High-throughput screening to identify inhibitors of lysine demethylases. Epigenomics 2015, 7, 57–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.; Zhang, P.; Yu, B. Advances toward LSD1 inhibitors for cancer therapy. Future Med. Chem. 2017, 9, 1227–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoumanne, A.; Chen, X. The lysine-specific demethylase 1 is required for cell proliferation in both p53-dependent and-independent manners. J. Biol. Chem. 2007, 282, 15471–15475. [Google Scholar] [CrossRef] [Green Version]
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586. [Google Scholar] [CrossRef]
- Huang, Y.; Stewart, T.M.; Wu, Y.; Baylin, S.B.; Marton, L.J.; Perkins, B.; Jones, R.J.; Woster, P.M.; Casero, R.A. Novel oligoamine analogues inhibit lysine-specific demethylase 1 and induce reexpression of epigenetically silenced genes. Clin. Cancer Res. 2009, 15, 7217–7228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, M.L.; Hauser, A.T.; Carlino, L.; Pippel, M.; Schulz-Fincke, J.; Metzger, E.; Willmann, D.; Yiu, T.; Barton, M.; Schule, R.; et al. Nonpeptidic propargylamines as inhibitors of lysine specific demethylase 1 (LSD1) with cellular activity. J. Med. Chem. 2013, 56, 7334–7342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, T.; Yuan, D.; Miao, X.; Lv, Y.; Zhan, P.; Shen, X.; Song, Y. Over-expression of LSD1 promotes proliferation, migration and invasion in non-small cell lung cancer. PLoS ONE 2012, 7, e35065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, T.E.; England, K.S.; Hopkinson, R.J.; Brennan, P.E.; Kawamura, A.; Schofield, C.J. Recent progress in histone demethylase inhibitors. J. Med. Chem. 2016, 59, 1308–1329. [Google Scholar] [CrossRef]
- Kruger, R.G.; Mohammad, H.; Smitheman, K.; Cusan, M.; Liu, Y.; Pappalardi, M.; Federowicz, K.; Van Aller, G.; Kasparec, J.; Tian, X.; et al. Inhibition of LSD1 as a therapeutic strategy for the treatment of acute myeloid leukemia. Blood 2013, 122, 3964. [Google Scholar] [CrossRef]
- Lee, S.H.; Stubbs, M.; Liu, X.M.; Diamond, M.; Dostalik, V.; Ye, M.; Lo, Y.; Favata, M.; Yang, G.; Gallagher, K.; et al. Abstract 4712: Discovery of INCB059872, a novel FAD-directed LSD1 inhibitor that is effective in preclinical models of human and murine AML. Cancer Res. 2016, 76, 4712. [Google Scholar]
- Milletti, F.; Cheng, W.Y.; Maes, T.; Lunardi, S.; DeMario, M.; Pierceall, W.E.; Mack, F. Abstract 4708: Neuroendocrine gene transcript expression is associated with efficacy to lysine-specific demethylase-1 inhibitor RG6016 in small cell lung cancer-derived cell lines. Cancer Res. 2016, 76, 4708. [Google Scholar]
- Gooden, D.M.; Schmidt, D.M.; Pollock, J.A.; Kabadi, A.M.; McCafferty, D.G. Facile synthesis of substituted trans-2-arylcyclopropylamine inhibitors of the human histone demethylase LSD1 and monoamine oxidases A and B. Bioorg. Med. Chem. Lett. 2008, 18, 3047–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willmann, D.; Lim, S.; Wetzel, S.; Metzger, E.; Jandausch, A.; Wilk, W.; Jung, M.; Forne, I.; Imhof, A.; Janzer, A.; et al. Impairment of prostate cancer cell growth by a selective and reversible lysine-specific demethylase 1 inhibitor. Int. J. Cancer 2012, 131, 2704–2709. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; He, Z.; Yang, M.; Gao, Y.; Jin, L.; Wang, M.; Zheng, Y.; Lu, X.; Zhang, S.; Wang, C.; et al. Investigating the Binding Mode of Reversible LSD1 Inhibitors Derived from Stilbene Derivatives by 3D-QSAR, Molecular Docking, and Molecular Dynamics Simulation. Molecules 2019, 24, 4479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; He, Z.; Liu, H.; Chen, Y.; Gao, Y.; Zhang, S.; Wang, M.; Lu, X.; Wang, C.; Zhao, Z.; et al. 3D-QSAR, molecular docking, and molecular dynamics simulation study of thieno [3, 2-b] pyrrole-5-carboxamide derivatives as LSD1 inhibitors. RSC Adv. 2020, 10, 6927–6943. [Google Scholar] [CrossRef]
- Abdizadeh, R.; Heidarian, E.; Hadizadeh, F.; Abdizadeh, T. QSAR modeling, molecular docking and molecular dynamics simulations studies of lysine-specific demethylase 1 (Lsd1) inhibitors as anticancer agents. Anti-Cancer Agents Med. Chem. 2021, 21, 987–1018. [Google Scholar] [CrossRef]
- Seraj, K.; Asadollahi-Baboli, M. In silico evaluation of 5-hydroxypyrazoles as LSD1 inhibitors based on molecular docking derived descriptors. J. Mol. Struct. 2019, 1179, 514–524. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Honório, K.M.; Emery, F.S.; Ganesan, A.; Trossini, G.H. Hologram quantitative structure–activity relationship and comparative molecular interaction field analysis of aminothiazole and thiazolesulfonamide as reversible LSD1 inhibitors. Future Med. Chem. 2015, 7, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Bergner, A.; Parel, S.P. Hit expansion approaches using multiple similarity methods and virtualized query structures. J. Chem. Inf. Model. 2013, 53, 1057–1066. [Google Scholar] [CrossRef]
- Zhou, C.; Kang, D.; Xu, Y.; Zhang, L.; Zha, X. Identification of Novel Selective Lysine-Specific Demethylase 1 (LSD1) Inhibitors Using a Pharmacophore-Based Virtual Screening Combined with Docking. Chem. Biol. Drug Des. 2015, 85, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.D.; Zheng, Y.C.; Ma, C.Y.; Yang, J.; Gao, Q.B.; Yan, Y.; Wang, Z.Z.; Li, W.; Zhao, W.; Liu, H.M.; et al. Identifying the novel inhibitors of lysine-specific demethylase 1 (LSD1) combining pharmacophore-based and structure-based virtual screening. J. Biomol. Struct. Dyn. 2018, 37, 4200–4214. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Chu, X.; Sun, X.Y.; Xu, K.; Deng, H.X.; Chen, J.; Wei, Z.; Lei, M. Machine learning in materials science. InfoMat 2019, 1, 338–358. [Google Scholar] [CrossRef]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N.O.; Baker, N. Chemception: A deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. arXiv 2017, arXiv:1706.06689. [Google Scholar]
- Li, L.; Hoyer, S.; Pederson, R.; Sun, R.; Cubuk, E.D.; Riley, P.; Burke, K. Kohn-Sham equations as regularizer: Building prior knowledge into machine-learned physics. Phys. Rev. Lett. 2021, 126, 036401. [Google Scholar] [CrossRef]
- Singh, S.; Pareek, M.; Changotra, A.; Banerjee, S.; Bhaskararao, B.; Balamurugan, P.; Sunoj, R.B. A unified machine-learning protocol for asymmetric catalysis as a proof of concept demonstration using asymmetric hydrogenation. Proc. Natl. Acad. Sci. USA 2020, 117, 1339–1345. [Google Scholar] [CrossRef]
- Haywood, A.L.; Redshaw, J.; Gaertner, T.; Taylor, A.; Mason, A.M.; Hirst, J.D. Machine Learning for Chemical Synthesis. In Machine Learning in Chemistry: The Impact of Artificial Intelligence; The Royal Society of Chemistry: London, UK, 2020; pp. 169–194. [Google Scholar]
- Melville, J.L.; Burke, E.K.; Hirst, J.D. Machine learning in virtual screening. Comb. Chem. High Throughput Scr. 2009, 12, 332–343. [Google Scholar] [CrossRef]
- Obermeyer, Z.; Emanuel, E.J. Predicting the future—Big data, machine learning, and clinical medicine. N. Engl. J. Med. 2016, 375, 1216. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comp. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandfort, F.; Strieth-Kalthoff, F.; Kühnemund, M.; Beecks, C.; Glorius, F. A structure-based platform for predicting chemical reactivity. Chem 2020, 6, 1379–1390. [Google Scholar] [CrossRef]
- Morgan, H.L. The generation of a unique machine description for chemical structures-a technique developed at chemical abstracts service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Haywood, A.L.; Redshaw, J.; Hanson-Heine, M.W.D.; Taylor, A.; Brown, A.; Mason, A.M.; Gaertner, T.; Hirst, J.D. Kernel Methods for Predicting Yields of Chemical Reactions. J. Chem. Inf. Model. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: New York, NY, USA, 2019; pp. 8024–8035. [Google Scholar]
- NVIDIA; Vingelmann, P.; Fitzek, F.H. CUDA, release: 10.2.89. 2020. Available online: https://developer.nvidia.com/cuda-toolkit (accessed on 9 November 2021).
- Sterling, T.; Irwin, J.J. ZINC 15–ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Hinton, G.; Roweis, S.T. Stochastic neighbor embedding. Adv. Neural Inf. Process. Syst. 2002, 15, 833–840. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Ricq, E.L.; Hooker, J.M.; Haggarty, S.J. Activity-dependent regulation of histone lysine demethylase KDM1A by a putative thiol/disulfide switch. J. Biol. Chem. 2016, 291, 24756–24767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Huang, H.; Zhang, Z.; Yan, J.; Wu, T.; Yin, W.; Sun, Y.; Wang, X.; Gu, Y.; Zhao, D.; et al. Design, synthesis and biological evaluation of novel benzofuran derivatives as potent LSD1 inhibitors. Eur. J. Med. Chem. 2021, 220, 113501. [Google Scholar] [CrossRef] [PubMed]
- Danqing, Z.; Xu, L.; Xiaojing, P.; Hongmin, L.; Qiurong, Z. Design, Synthesis and Anticancer Activity Studies of Novel Indole-Pyrimidine Biaryl Derivatives. Chin. J. Org. Chem. 2021, 41, 267–275. [Google Scholar]
- Niwa, H.; Sato, S.; Hashimoto, T.; Matsuno, K.; Umehara, T. Crystal Structure of LSD1 in Complex with 4-[5-(Piperidin-4-ylmethoxy)-2-(p-tolyl) pyridin-3-yl] benzonitrile. Molecules 2018, 23, 1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.J.; Liu, Y.; Xue, L.P.; Xiong, X.P.; Zhou, Y.; Zheng, Y.C.; Liu, H.M. Reversible Lysine Specific Demethylase 1 (LSD1) Inhibitors: A Promising Wrench to Impair LSD1. J. Med. Chem. 2021, 64, 2466–2488. [Google Scholar] [CrossRef]
- Xi, J.; Xu, S.; Wu, L.; Ma, T.; Liu, R.; Liu, Y.C.; Deng, D.; Gu, Y.; Zhou, J.; Lan, F. Design, synthesis and biological activity of 3-oxoamino-benzenesulfonamides as selective and reversible LSD1 inhibitors. Bioorg. Chem. 2017, 72, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Shi, L.; Lai, C.; Severin, C.; Xu, J.; Del Rosario, J.R.; Stansfield, R.K.; Cho, R.W.; Kanouni, T.; Veal, J.M.; et al. Structure-based design and discovery of potent and selective lysine-specific demethylase 1 (LSD1) inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 103–106. [Google Scholar] [CrossRef]
- Neelamegam, R.; Ricq, E.L.; Malvaez, M.; Patnaik, D.; Norton, S.; Carlin, S.M.; Hill, I.T.; Wood, M.A.; Haggarty, S.J.; Hooker, J.M. Brain-penetrant LSD1 inhibitors can block memory consolidation. ACS Chem. Neurosci. 2012, 3, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, K.G.; Sudalai, A. A Concise Enantioselective Synthesis of (+)-L-733,060 and (+)-T-2328 via Sequential Proline Catalysis. Synlett 2016, 27, 1339–1343. [Google Scholar] [CrossRef]
- Kuczynski, W.; Pierzynski, P. Use of Antagonist of Oxytocin and/or Vasopressin in Assisted Reproduction. U.S. Patent Application 11/914,049, 25 December 2008. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Algorithm | Train | Test | Train RMSE | Test RMSE |
|---|---|---|---|---|
| K-Neighbours | 0.998 (0.001) | 0.662 (0.047) | 0.051 (0.010) | 0.632 (0.051) |
| Ridge | 0.923 (0.005) | 0.471 (0.069) | 0.306 (0.011) | 0.790 (0.059) |
| Lasso | 0.688 (0.009) | 0.597 (0.044) | 0.616 (0.010) | 0.690 (0.044) |
| Elastic Net | 0.821 (0.006) | 0.635 (0.047) | 0.466 (0.009) | 0.656 (0.047) |
| Gradient Boosting | 0.833 (0.007) | 0.631 (0.041) | 0.450 (0.010) | 0.661 (0.040) |
| Random Forest | 0.984 (0.001) | 0.695 (0.035) | 0.140 (0.004) | 0.600 (0.041) |
| Adaboost | 0.582 (0.017) | 0.500 (0.034) | 0.713 (0.015) | 0.769 (0.035) |
| Extra Trees | 0.998 (0.001) | 0.459 (0.092) | 0.051 (0.010) | 0.798 (0.073) |
| Decision tree | 0.931 (0.009) | 0.425 (0.090) | 0.288 (0.020) | 0.823 (0.066) |
| SVR | 0.989 (0.001) | 0.703 (0.035) | 0.115 (0.005) | 0.592 (0.041) |
| MLP | 0.998 (0.001) | 0.544 (0.218) | 0.052 (0.010) | 0.723 (0.127) |
| Dataset | Algorithm | Test | Test RMSE |
|---|---|---|---|
| Subset 1 a | RF | 0.498 (0.172) | 0.651 (0.106) |
| SVR | 0.536 (0.189) | 0.623 (0.117) | |
| DT | 0.292 (0.247) | 0.772 (0.124) | |
| Subset 2 b | RF | 0.760 (0.055) | 0.499 (0.057) |
| SVR | 0.745 (0.055) | 0.516 (0.053) | |
| DT | 0.515 (0.133) | 0.710 (0.107) | |
| Subset 3 c | Ridge | 0.670 (0.141) | 0.509 (0.054) |
| SVR | 0.662 (0.143) | 0.516 (0.062) | |
| DT | 0.379 (0.253) | 0.701 (0.108) | |
| Subset 4 d | RF | 0.458 (0.069) | 0.654 (0.053) |
| SVR | 0.473 (0.081) | 0.646 (0.069) | |
| DT | 0.112 (0.171) | 0.833 (0.069) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, J.; Wu, S.; Lee, B.G.; Chen, T.; He, Z.; Lei, Y.; Tang, B.; Hirst, J.D. Machine-Learning-Enabled Virtual Screening for Inhibitors of Lysine-Specific Histone Demethylase 1. Molecules 2021, 26, 7492. https://doi.org/10.3390/molecules26247492
Zhou J, Wu S, Lee BG, Chen T, He Z, Lei Y, Tang B, Hirst JD. Machine-Learning-Enabled Virtual Screening for Inhibitors of Lysine-Specific Histone Demethylase 1. Molecules. 2021; 26(24):7492. https://doi.org/10.3390/molecules26247492
Chicago/Turabian StyleZhou, Jiajun, Shiying Wu, Boon Giin Lee, Tianwei Chen, Ziqi He, Yukun Lei, Bencan Tang, and Jonathan D. Hirst. 2021. "Machine-Learning-Enabled Virtual Screening for Inhibitors of Lysine-Specific Histone Demethylase 1" Molecules 26, no. 24: 7492. https://doi.org/10.3390/molecules26247492
APA StyleZhou, J., Wu, S., Lee, B. G., Chen, T., He, Z., Lei, Y., Tang, B., & Hirst, J. D. (2021). Machine-Learning-Enabled Virtual Screening for Inhibitors of Lysine-Specific Histone Demethylase 1. Molecules, 26(24), 7492. https://doi.org/10.3390/molecules26247492