3.2. Reagents
N,
N′-Carbonyldiimidazole (CDI), tryptamine, NaH (60% in mineral oil), Lawesson’s reagent, and 4-bromophenylacetonitrile were purchased from Acros Organics (Acros Organics B.V.B.A., Geel, Belgium). Benzyl bromide was purchased from Alfa Aesar. 3-Acetylaniline and
n-Bu
4NF were purchased from Sigma Aldrich. 4-Acetylaniline was purchased from J&K. Deoxycholic acid (DCA) was purchased from Abcr GmbH & Co. KG. Dimethyl sulfate was purchased from VEKTON. 4-Bromobenzonitrile and 4-bromobenzaldehyde were purchased from Fluorochem. All solvents used in the reactions were purified and dried according to previously reported procedures. 4-Bromo
-N′-hydroxybenzimidamide and 2-(4-bromophenyl)-
N′-hydroxyacetimidamide were prepared according to the method in the literature [
27].
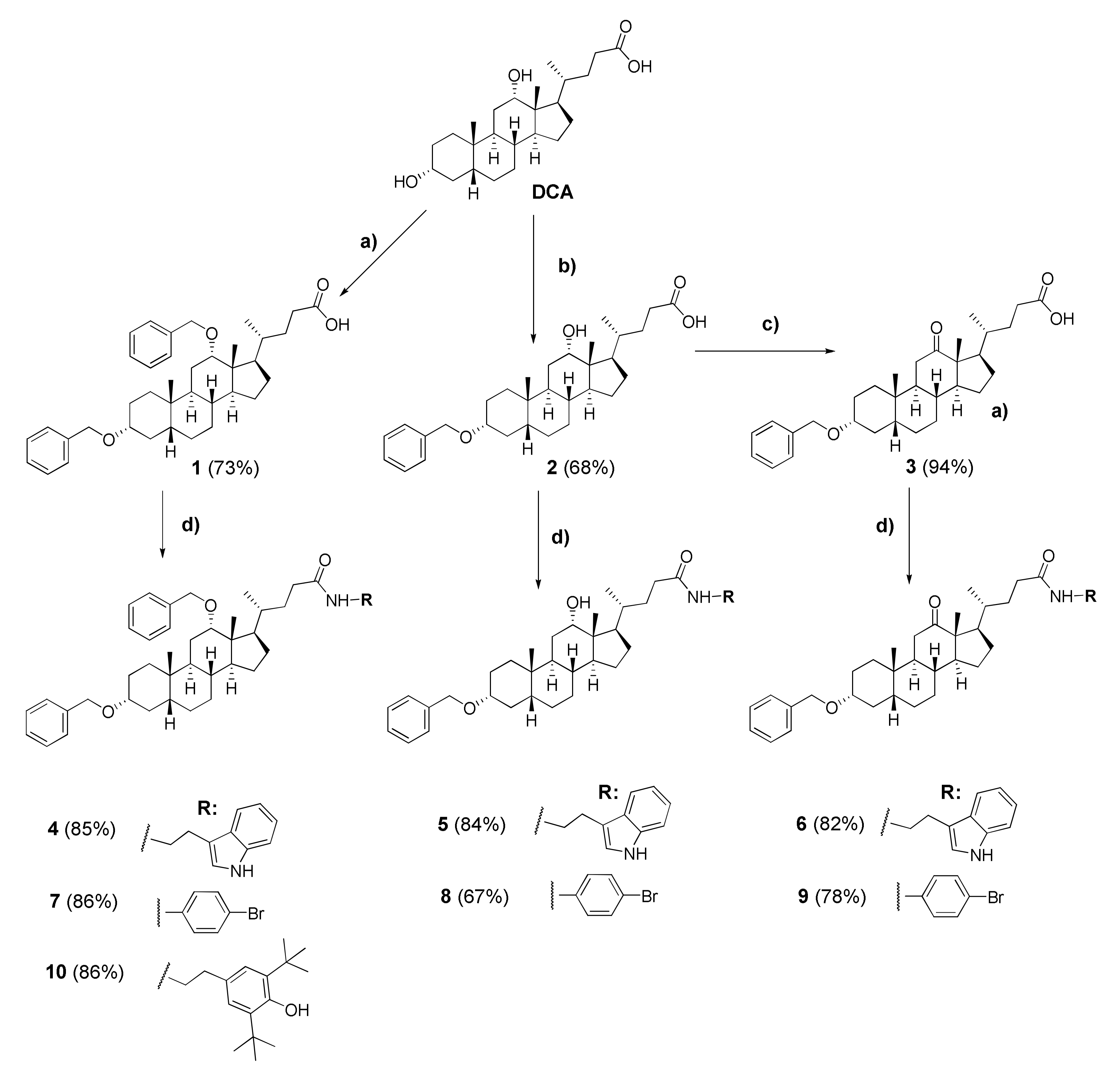
3.2.1. 3α,12α-Dibenzyloxy-5β-cholan-24-oic acid (1)
Mixture of DCA (0.6 g, 1.53 mmol) and NaH (57–63% in oil; 0.37 g, 9.18 mmol) in THF (10 mL) was heated at 50 °C for 1 h. Then, benzyl bromide (0.56 mL, 4.59 mmol) was added, and the reaction mixture was refluxed for 24 h. Next, NaH (57–63% in oil; 0.18 g, 4.60 mmol) and benzyl bromide (0.27 mL, 2.30 mmol) were added to the reaction mixture, and the reaction mixture was refluxed until full conversion was reached. The reaction course was monitored by TLC. The reaction mixture was cooled to room temperature, concentrated under vacuum, diluted with AcOEt–CHCl3 mixture, washed with aqueous NH4Cl, and dried over MgSO4. Crude product (1.27 g) was purified by flash column chromatography (SiO2, CH2Cl2 then CHCl3) to give a pure sample of compound 1 (0.64 g, 73%) as a white amorphous solid. Mp 181.2 °C [decomposition]. [] +60 (c 0.20 g/100 mL; CHCl3). HRMS: Calc. for (C38H52O4)+ m/z = 572.3860; found m/z = 481.3314; calc. for (C31H45O4)+ m/z = 481.3312 [M–PhCH2]+. 1H NMR (CDCl3, 500 MHz): δ = 7.38–7.20 (m, 10H, aromatic protons), 4.59 (d, 1H, J = 11.4, H-26), 4.52 (s, 2H, CH2-25), 4.59 (d, 1H, J = 11.4, H-26′), 3.66 (s, 1H, H-12), 3.35 (m, 1H, H-3), 2.38 (m, 1H, H-23), 2.21 (m, 1H, H-23′), 2.02 (ddd, 1H, J = J = J = 9.6, H-17), 1.89–1.66 (m, 9H; H-4, H-9, H-20, H-6, H-22, H-14, 3H), 1.64–1.50 (m, 2H, H-4, H-15), 1.47–1.18 (m, 9H,H-8, H-5, H-22′, H-6′, H-7, 4H), 1.13 (m, 1H, H-7′), 1.03 (m, 1H, H-15′), 0.93 (m, 1H, H-1′), 0.91 (s, 3H, CH3-19), 0.87 (d, 3H, J21,20 = 6.3, CH3-21), 0.69 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 179.51 (s, C-24), 139.24 (s, C-1Ph *), 139.17 (s, C-1Ph′ *), 128.16 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 127.45 (d, C-2Ph, C-6Ph, C-2Ph′, C-6Ph′), 127.37 (d, C-4Ph #), 127.14 (d, C-4Ph′ #), 80.91 (d, C-12), 78.54 (d, C-3), 70.20 (t, C-26), 69.50 (t, C-25), 48.64 (d, C-14), 46.54 (s, C-13), 46.13 (d, C-17), 42.17 (d, C-5), 36.06 (d, C-8), 34.29 (s, C-10§), 35.15 (d, C-20), 34.47 (t, C-1§), 33.74 (d, C-9), 33.20 (t, C-4), 30.75 (t, C-23), 30.70 (t, C-22), 27.46, (t, C-11‡), 27.31 (t, C-16‡), 27.21 (t, C-2‡), 25.97 (t, C-7), 23.64 (t, C-15), 23.26 (q, C-19), 23.02 (t, C-6), 17.37 (q, C-21), 12.66 (q, C-18).
3.2.2. 3α-Benzyloxy-12α-hydroxy-5β-cholan-24-oic Acid (2)
Mixture of DCA (1.0 g, 2.55 mmol) and NaH (57–63% in oil; 0.5 g, 11.9 mmol) in THF (20 mL) was heated at 50 °C for 3 h. Then, benzyl bromide (0.92 mL, 7.65 mmol) was added, and the reaction mixture was refluxed until full conversion was reached. The reaction course was monitored by TLC. The reaction mixture was cooled to room temperature, concentrated under vacuum, diluted with AcOEt–CHCl3 mixture, washed with aqueous NH4Cl, and dried over MgSO4. The crude product (1.9 g) was purified by flash column chromatography (SiO2, CHCl3) to give a pure sample of compound 2 (0.84 g, 68%) as a white amorphous solid. Mp 86.0 °C [decomposition]. [] +47 (c 0.20 g/100 mL; CHCl3). HRMS: m/z calcd. for (C31H46O4)+ 482.3391; found 482.3379. 1H NMR (CDCl3, 500 MHz): δ = 7.34–7.28 (m, 4H, H-2Ph, H-3Ph, H-5Ph, H-6Ph), 7.25 (m, 1H, H-4Ph), 4.55 (s, 2H, CH2-25), 3.92 (s, 1H, H-12), 3.39 (m, 1H, H-3), 2.40 (m, 1H, H-23), 2.18 (m, 1H, H-23′), 2.00–1.89 (m, 2H; H-4, H-9), 1.89–1.68 (m, 6H, H-16, H-6, H-17, H-22, H-1, H-2), 1.64–1.46 (m, 6H, H-4′, H-15, H-2′, H-14, CH2-11), 1.43–1.27 (m, 5H, H-8, H-7, H-22′, H-20, H-5), 1.27–1.00 (m, 4H, H-6′, H-16′, H-7′, H-15′), 0.96 (d, 3H, J21,20 = 6.2, CH3-21), 0.87 (s, 3H, CH3-19), 0.87 (m, 1H, H-1′), 0.66 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 178.43 (s, C-24), 138.63 (s, C-1Ph), 128.22 (d, C-3Ph, C-5Ph), 127.52 (d, C-2Ph, C-6Ph), 127.36 (d, C-4Ph), 78.85 (d, C-3), 73.17 (d, C-12), 69.80 (t, C-25), 48.30 (d, C-14), 47.50 (d, C-17), 46.52 (s, C-13), 42.04 (d, C-5), 35.85 (d, C-8), 35.46 (d, C-20), 35.19 (t, C-1), 34.30 (s, C-10), 33.14 (d, C-9), 32.69 (t, C-4), 31.07 (t, C-23), 30.67 (t, C-22), 28.46, (t, C-11), 27.56 (t, C-16), 27.05 (t, C-6), 26.60 (t, C-2), 25.94 (t, C-7), 23.60 (t, C-15), 22.81 (q, C-19), 17.04 (q, C-21), 12.60 (q, C-18).
3.2.3. 3α-Benzyloxy-12α-oxo-5β-cholan-24-oic Acid (3)
To a solution of compound 2 (4.6 g, 9.5 mmol) in acetone (150 mL) Jones reagent (3 mL) was added dropwise. The reaction mixture was stirred for 2 h at room temperature. The reaction course was monitored by TLC. Then, EtOH (10 mL) was added and the reaction mixture was stirred for an extra 30 min. Precipitate was filtered, organic phase was concentrated under vacuum, diluted with water, extracted with CH2Cl2–Et2O, and dried over anhydrous MgSO4. Crude product (4.47 g, 98%) was purified by flash column chromatography (SiO2, 0–1% MeOH gradient in CHCl3) to give a pure sample of 3 (4.3 g, 94%). Mp 147.9°C (AcOEt) [decomposition]. [] +90 (c 0.20 g/100 mL; CHCl3). HRMS: m/z calcd. for (C31H44O4)+ 480.3234; found 480.3231. 1H NMR (CDCl3, 500 MHz): δ = 7.32–7.28 (m, 4H, H-2Ph, H-3Ph, H-5Ph, H-6Ph), 7.26–7.21 (m, 1H, H-4Ph), 4.52 (ddd, 2H, J = J = J = 10.7, CH2-25), 3.34 (m, 1H, H-3), 2.43 (dd, 1H, J = J = 12.5, H-11β), 2.41 (m, 1H, H-23), 2.27 (m, 1H, H-23′), 2.07–1.22 (m, 21H), 1.11 (m, 1H, H-7), 1.02–0.96 (m, 7H; H-1, 0.99 (s, 3H, CH3-18*), 0.98 (s, 3H, CH3-19*)), 0.84 (d, 3H, J = 6.6, CH3-21). 13C NMR (CDCl3, 125 MHz): δ = 214.49 (s, C-12), 179.30 (s, C-24), 138.94 (s, C-1Ph), 128.22 (d, C-3Ph, C-5Ph), 127.41 (d, C-2Ph, C-6Ph), 127.25 (d, C-4Ph), 77.95 (d, C-3), 69.69 (t, C-25), 58.44 (d, C-14), 57.38 (s, C-13), 46.35 (d, C-17), 43.88 (d, C-9), 41.52 (d, C-5), 37.97 (t, C-11), 35.62, 35.57, 35.50, 35.16, 33.16, 31.09, 30.20, 27.41, 27.13, 26.77, 25.94 (t, C-7), 24.2 (t, C-15), 22.71 (q, C-19), 18.44 (q, C-21), 11.59 (q, C-18).
3.2.4. N-(2″-(1H-Indol-3′-yl)ethyl)-3α,12α-dibenzyloxy-5β-cholane-24-amide (4)
Compound 1 (0.30 g, 0.52 mmol) and CDI (0.10 g, 0.62 mmol) was dissolved in CH2Cl2 (10 mL) and stirred at room temperature for 2 h. Then, tryptamine (0.10 g, 0.62 mmol) was added and the reaction mixture was stirred at 30–35 °C for 8 h. The reaction course was monitored by TLC (CHCl3). Reaction mixture was diluted with CH2Cl2 and Et2O, washed sequentially with H2O and brine, and dried over anhydrous MgSO4. Solvent was evaporated to dryness; crude product (0.40 g, quantitative yield) was purified by flash column chromatography (SiO2, n-hexane with gradient 10–50% AcOEt) to give a pure sample of compound 4 (0.32 g, 85%) as a white amorphous solid. Mp 72.0 °C [decomposition]. [] +52 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C48H62O3N2)+ m/z = 714.4755; found m/z = 714.4748. 1H-NMR (CDCl3, 500 MHz): δ = 8.16 (s, NH, indole), 7.59 (d, 1H, J = 7.7, H-5′), 7.41–7.06 (m, 13H, aromatic protons of Ph and Ph′, H-8′, H-7′, H-6′), 6.99 (br.s., 1H, H-2′), 5.43 (m, NH, amide), 4.59 (d, 1H, J = 11.4, H-26), 4.52 (m, CH2-25), 4.25 (d, 1H, J = 11.4, H-26′), 3.65 (s, 1H, H-12), 3.56 (m, 2H, CH2-1″), 3.33 (m, 1H, H-3), 2.94 (m, CH2-2″), 2.13 (m, 1H, H-23), 2.06–0.78 (m, 31H; 0.91 (s, 3H, CH3-19), 0.84 (d, 3H, J = 6.2, CH3-21)), 0.67 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 173.47 (s, C-24), 139.22 (s, C-1Ph *), 139.19 (s, C-1Ph′ *), 136.33 (s, C-9^), 128.16 (d, C-3Ph, C-5Ph #), 128.13 (C-3Ph′, C-5Ph′ #), 127.42 (d, C-2Ph, C-6Ph ∞), 127.36 (d, C-2Ph′, C-6Ph′ ∞), 127.28 (s, C-4^), 127.15 (d, C-4Ph §), 127.09 (d, C-4Ph′ §), 122.04 (d, C-2^), 121.87 (d, C-7^), 119.34 (d, C-6^), 118.60 (d, C-5^), 113.00 (s, C-3^), 111.13 (d, C-8^), 80.97 (d, C-12), 78.51 (d, C-3), 70.16 (t, C-26), 69.52 (t, C-25), 48.68 (d, C-14), 46.46 (s, C-13), 46.03 (d, C-17), 42.11 (d, C-5), 39.57 (t, C-1″), 35.99 (d, C-8), 35.26 (d, C-20), 35.23 (s, C-10‡), 34.43 (t, C-1‡), 33.72 (d, C-9), 34.37 (t, C-23), 33.19 (t, C-4), 31.64 (t, C-22), 27.50 (t, C-11†), 27.26 (t, C-16†), 27.19 (t, C-2†), 25.94 (t, C-7), 25.25 (t, C-2″), 23.61 (t, C-15), 23.25 (q, C-19), 22.97 (t, C-6), 17.46 (q, C-21), 12.62 (q, C-18).
3.2.5. N-(2″-(1H-Indol-3′-yl)ethyl)-3α-benzyloxy-12α-hydroxy-5β-cholane-24-amide (5)
Compound 2 (0.25 g, 0.52 mmol) and CDI (0.10 g, 0.62 mmol) was dissolved in CH2Cl2 (10 mL) and stirred at room temperature for 2 h. Then, tryptamine (0.10 g, 0.62 mmol) was added and reaction mixture was stirred at 30–35 °C for 36 h. The reaction course was monitored by TLC (CHCl3–AcOEt, 15:10). Reaction mixture was diluted with CH2Cl2 and Et2O, washed sequentially with H2O, HCl 5% (aq), NaHCO3 (aq), brine, and dried over anhydrous MgSO4. Solvent was evaporated to dryness; crude product (0.35 g, quantitative yield) was purified by flash column chromatography (SiO2, CH2Cl2 with gradient 10–30% AcOEt) to give a pure sample of compound 5 (0.27 g, 84%) as a white amorphous solid. Mp 81.7 °C [decomposition]. [] +40 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C41H56O3N2)+ m/z = 624.4286; found m/z = 624.4285. 1H-NMR (CDCl3, 300 MHz): δ = 8.54 (br.s., NH, indole), 7.63–7.53 (d, 1H, J = 7.7, H-5^), 7.42–7.21 (m, 5H, H-2Ph, H-3Ph, H-4Ph, H-5Ph, H-6Ph), 7.21–7.04 (m, 2H, H-7^, H-6^), 6.99 (m, 1H, H-2^), 5.69 (m, NH, amide), 4.55 (s, 2H, CH2-25), 3.89 (s, 1H, H-12), 3.56 (m, 2H, CH2-1″), 3.37 (m, 1H, H-3), 2.94 (m, 2H, H-2″), 2.14–0.79 (m, 33H, 2.14 (m, 1H, H-23), 1.97 (m, 1H, H-23′), 0.90 (d, 3H, J = 6.2, CH3-21), 0.88 (s, 3H, CH3-19)), 0.62 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 173.54 (s, C-24), 138.85 (s, C-1Ph), 136.25 (s, C-9^), 128.21 (d, C-3Ph, C-5Ph), 127.45 (d, C-2Ph, C-6Ph), 127.26 (d, C-4Ph), 127.17 (s, C-4^), 122.06 (d, C-2^), 121.86 (d, C-7^), 119.16 (d, C-6^), 118.50 (d, C-5^), 112.68 (s, C-3^), 111.22 (d, C-8^), 78.44 (d, C-3), 72.92 (d, C-12), 69.72 (t, C-25), 48.05 (d, C-14), 46.83 (d, C-17), 46.24 (s, C-13), 41.85 (d, C-5), 39.50 (t, C-1″), 35.78 (d, C-8), 35.00 (s, C-10*), 34.96 (d, C-20), 34.24 (t, C-1*), 33.43 (d, C-9), 33.26 (t, C-23#), 32.98 (t, C-4#), 31.45 (t, C-22), 28.48 (t, C-11), 27.25 (t, C-16§), 27.03 (t, C-6§), 26.91 (t, C-2§), 25.87 (t, C-7), 25.14 (t, C-2″), 23.45 (t, C-15), 23.02 (q, C-19), 17.20 (q, C-21), 12.60 (q, C-18).
3.2.6. N-(2″-(1H-Indol-3′-yl)ethyl)-3α-benzyloxy-12-oxo-5β-cholane-24-amide (6)
Compound 3 (0.3 g, 0.62 mmol) and CDI (0.12 g, 0.75 mmol) was dissolved in CH2Cl2 (10 mL) and stirred at room temperature for 2 h. Then, tryptamine (0.12 g, 0.75 mmol) was added and the reaction mixture was stirred at 30–35 °C overnight. The reaction course was monitored by TLC (CHCl3–AcOEt, 20:3). Reaction mixture was diluted with CH2Cl2 and Et2O, washed sequentially with H2O and brine, and dried over anhydrous MgSO4. Solvent was evaporated to dryness; crude product (0.42 g, quantitative yield) was purified by flash column chromatography (SiO2, n-hexane with gradient 20–50% AcOEt) to give a pure sample of compound 6 (0.32 g, 82%) as a white amorphous solid. Mp 87.5 °C [decomposition]. [] +70 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C41H54O3N2)+ m/z = 622.4129; found m/z = 622.4124. 1H-NMR (CDCl3, 500 MHz): δ = 8.42 (s, NH, indole), 7.62–7.54 (m, 1H, H-5^), 7.37–7.21 (m, 6H, H-8^, H-2Ph, H-3Ph, H-4Ph, H-5Ph, H-6Ph), 7.17 (m, 1H, H-7^), 7.09 (m, 1H, H-6^), 6.99 (br.s., 1H, H-2^), 5.62 (m, NH, amide), 4.52 (ddd, 2H, J = J = J = 10.2, CH2-25), 3.57 (m, 2H, CH2-1″), 3.35 (m, 1H, H-3), 2.95 (m, 2H, CH2-2″), 2.43 (dd, 1H, J = J = 12.5, H-11β), 2.17 (m, 1H, H-23), 2.08–0.89 (m, 30H, 0.98 (s, 3H, CH3-18*), 0.96 (s, 3H, CH3-19*)), 0.80 (d, 3H, J = 6.5, CH3-21). 13C NMR (CDCl3, 125 MHz): δ = 214.65 (s, C-12), 173.30 (s, C-24), 138.88 (s, C-1Ph), 136.33 (s, C-9^), 128.19 (d, C-3Ph, C-5Ph), 127.37 (d, C-2Ph, C-6Ph), 127.24 (d, C-4Ph), 121.92 (d, C-2^, C-7^), 119.21 (d, C-6^), 118.53 (d, C-5^), 112.81 (s, C-3^), 111.16 (d, C-8^), 77.92 (d, C-3), 69.69 (t, C-25), 58.44 (d, C-14), 57.34 (s, C-13), 46.22 (d, C-17), 43.87 (d, C-9), 41.44 (d, C-5), 39.55 (t, C-1″), 37.95 (t, C-11), 35.52 (d, C-8), 35.49 (s, C-10§), 35.43 (d, C-20), 35.09 (t, C-1§), 33.56 (t, C-23), 33.10 (t, C-4), 31.02 (t, C-22), 27.29, 27.05, 26.74, 25.87 (t, C-7), 25.22 (t, C-2″), 24.15, 22.64 (q, C-19), 18.59 (q, C-21), 11.55 (q, C-18).
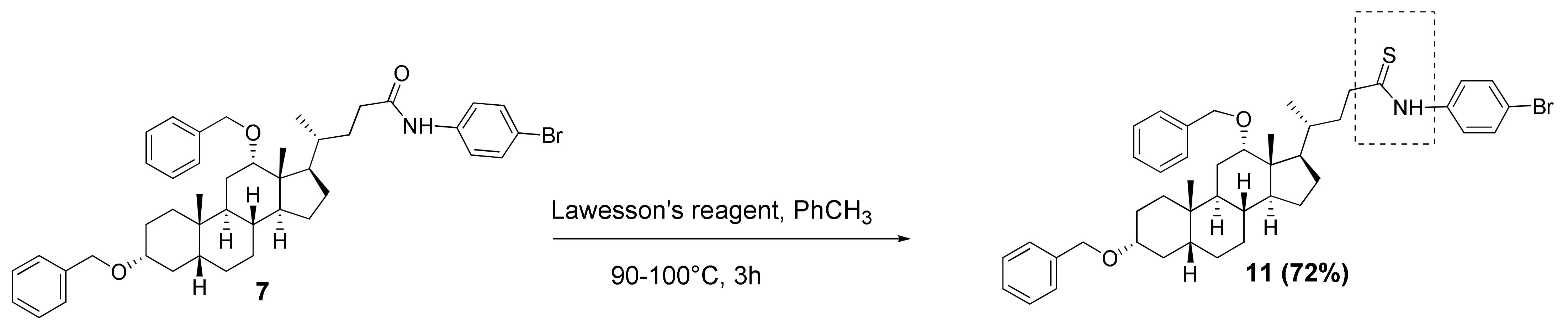
3.2.7. N-(4′-Bromophenyl)-3α,12α-dibenzyloxy-5β-cholane-24-amide (7)
Compound 1 (0.39 g, 0.68 mmol) and CDI (0.33 g, 2.0 mmol) was dissolved in CH2Cl2 (10 mL) and stirred at room temperature for 2 h, then the solution was heated to 30–35 °C and stirred 1 h more. Next, 4-bromoaniline (0.15 g, 0.89 mmol) was added and the reaction mixture was stirred at 30–35 °C for 2 h. The reaction course was monitored by TLC. Reaction mixture was diluted with AcOEt, washed sequentially with H2O, HCl 5% (aq), NaHCO3 (aq), brine, and dried over anhydrous MgSO4. Solvent was evaporated to dryness; crude product (0.52 g, quantitative yield) was purified by flash column chromatography (SiO2, CH2Cl2) to give a pure sample of compound 7 (0.42 g, 86%) as a white amorphous solid. Mp 186.2–191.3 °C. [] +20 (c 0.20 g/100 mL; CHCl3). HRMS: Calc. for (C44H56O3N79Br)+ m/z = 725.3438; found m/z = 725.3444. 1H-NMR (CDCl3, 400 MHz): δ = 7.44–7.21 (m, 15H, all aromatic protons, NH), 4.61 (d, 1H, J = 11.5, H-26), 4.52 (s, CH2-25), 4.27 (d, 1H, J = 11.5, H-26′), 3.67 (s, 1H, H-12), 3.36 (m, 1H, H-3), 2.33 (m, 1H, H-23), 2.16 (m, 1H, H-23′), 2.02 (m, 1H, H-17), 1.91–0.83 (m, 29H, 0.92 (s, 3H, CH3-19), 0.89 (d, 3H, J = 5.4, CH3-21)), 0.69 (s, 3H, CH3-18). 13C NMR (CDCl3, 100 MHz): δ = 171.94 (s, C-24), 139.18 (s, C-1Ph *), 139.02 (s, C-1Ph′ *), 136.94 (s, C-1^), 131.68 (d, C-3^, C-5^), 128.13 (d, C-3Ph, C-5Ph #), 128.11 (C-3Ph′, C-5Ph′ #), 127.33 (d, C-2Ph, C-6Ph, C-2Ph′, C-6Ph′), 127.14 (d, C-4Ph §), 127.07 (d, C-4Ph′ §), 121.20 (d, C-2^, C-6^), 116.40 (s, C-4^), 80.88 (d, C-12), 78.42 (d, C-3), 70.03 (t, C-26), 69.46 (t, C-25), 48.62 (d, C-14), 46.39 (s, C-13), 45.90 (d, C-17), 41.99 (d, C-5), 35.89 (d, C-8), 35.14 (s, C-10‡), 35.09 (d, C-20), 34.36 (t, C-1‡), 34.05 (t, C-23†), 33.62 (d, C-9), 33.08 (t, C-4), 31.26 (t, C-22†), 27.46, 27.31, 27.21, 25.87 (t, C-7), 23.55 (t, C-15), 23.22 (q, C-19), 22.86 (t, C-6), 17.43 (q, C-21), 12.61 (q, C-18).
3.2.8. N-(4′-Bromophenyl)-3α-benzyloxy-12α-hydroxy-5β-cholane-24-amide (8)
Compound 2 (0.35 g, 0.73 mmol) and CDI (0.35 g, 2.2 mmol) was dissolved in CH2Cl2 (10 mL) and stirred at room temperature for 2 h, then, the solution was heated to 30–35 °C and stirred 1 h more. Next, 4-bromoaniline (0.20 g, 1.16 mmol) was added and the reaction mixture was stirred at 30–35 °C for 2 h. The reaction course was monitored by TLC. The reaction mixture was diluted with AcOEt, washed sequentially with H2O, HCl 5% (aq), NaHCO3 (aq), brine, and dried over anhydrous MgSO4. Solvent was evaporated to dryness; crude product (0.44 g, 96%) was purified by flash column chromatography (SiO2, CH2Cl2 with gradient 0–1.0% MeOH) to give compound 8 (0.08 g) and a pure sample of compound 8 (0.24 g, 67%) as a white amorphous solid. Mp 95.6–102.3 °C. [] +29 (c 0.20 g/100 mL; CHCl3). HRMS: Calc. for (C37H50O3N79Br)+ m/z = 635.2969; found m/z = 635.2963. 1H-NMR (CDCl3, 500 MHz): δ = 7.58 (br.s., 1H, NH), 7.42–7.35 (m, 4H, H-2^, H-3^, H-5^, H-6^), 7.33–7.28 (m, 4H, H-2Ph, H-3Ph, H-5Ph, H-6Ph), 7.27–7.21 (m, 1H, H-4Ph), 4.53 (s, 2H, CH2-25), 3.93 (s, 1H, H-12), 3.36 (m, 1H, H-3), 2.36 (m, 1H, H-23), 2.21 (m, 1H, H-23′), 2.00–0.83 (m, 31H; 0.96 (d, 3H, J = 5.9, CH3-21), 0.88 (s, 3H, CH3-19)), 0.64 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 171.92 (s, C-24), 138.81 (s, C-1Ph), 137.04 (s, C-1^), 131.72 (d, C-3^, C-5^), 128.22 (d, C-3Ph, C-5Ph), 127.47 (d, C-2Ph, C-6Ph), 127.29 (d, C-4Ph), 121.20 (d, C-2^, C-6^), 116.39 (s, C-4^), 78.42 (d, C-3), 72.99 (d, C-12), 69.73 (t, C-25), 48.10 (d, C-14), 46.76 (d, C-17), 46.30 (s, C-13), 41.89 (d, C-5), 35.83 (d, C-8), 35.01, 34.90 (d, C-20), 34.29, 34.03, 33.47, 33.00, 31.15, 28.61, 27.31, 27.06, 26.94, 25.92 (t, C-7), 23.49 (t, C-15), 23.06 (q, C-19), 17.30 (q, C-21), 12.64 (q, C-18).
3.2.9. N-(4′-Bromophenyl)-3α-benzyloxy-12-oxo-5β-cholane-24-amide (9)
Compound 3 (1.0 g, 2.08 mmol) and CDI (0.4 g, 2.5 mmol) was dissolved in CH2Cl2 (15 mL) and stirred at room temperature for 2 h, then, the solution was heated to 30–35 °C and stirred 1 h more. Next, 4-bromoaniline (0.43 g, 2.5 mmol) was added and the reaction mixture was stirred at 30–35 °C overnight. The reaction course was monitored by TLC. Reaction mixture was diluted with AcOEt, washed sequentially with H2O, HCl 5% (aq), NaHCO3 (aq), brine, and dried over anhydrous MgSO4. Solvent was evaporated to dryness; crude product (1.38 g, quantitative yield) was purified by flash column chromatography (SiO2, CH2Cl2) to give a pure sample of compound 9 (1.03 g, 78%) as a white amorphous solid. Mp 90.2 °C [decomposition]. [] +58 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C37H48O3N79Br)+ m/z = 633.2812; found m/z = 633.2824. 1H-NMR (CDCl3, 500 MHz): δ = 7.43–7.37 (m, 4H, H-2^, H-3^, H-5^, H-6^), 7.36–7.28 (m, 5H, NH, H-2Ph, H-3Ph, H-5Ph, H-6Ph), 7.27–7.21 (m, 1H, H-4Ph), 4.52 (ddd, 2H, J = J = J = 10.4, CH2-25), 3.34 (m, 1H, H-3), 2.44 (dd, 1H, J = J = 12.6, H-11β), 2.41 (m, 1H, H-23), 2.27 (m, 1H, H-23′), 2.07–1.98 (m, 2H), 1.97–1.58 (m, 10H), 1.52–1.39 (m, 3H), 1.39–1.26 (m, 6H), 1.10 (m, 1H, H-7), 1.03–0.92 (m, 7H, H-1; 0.99 (s, 3H, CH3-18*), 0.98 (s, 3H, CH3-19*)), 0.87 (d, 3H, J = 6.6, CH3-21). 13C NMR (CDCl3, 125 MHz): δ = 214.73 (s, C-12), 171.62 (s, C-24), 138.93 (s, C-1Ph #), 137.06 (s, C-1^ #), 131.80 (d, C-3^, C-5^), 128.23 (d, C-3Ph, C-5Ph), 127.41 (d, C-2Ph, C-6Ph), 127.27 (d, C-4Ph), 121.21 (d, C-2^, C-6^), 116.47 (s, C-4^), 77.92 (d, C-3), 69.73 (t, C-25), 58.54 (d, C-14), 57.42 (s, C-13), 46.13 (d, C-17), 43.94 (d, C-9), 41.49 (d, C-5), 38.00 (t, C-11), 35.59 (d, C-8), 35.56 (s, C-10§), 35.40 (d, C-20), 35.14 (t, C-1§), 34.39 (t, C-23), 33.15 (t, C-4), 30.74 (t, C-22), 27.42, 27.10, 26.79, 25.93 (t, C-7), 24.22, 22.70 (q, C-19), 18.64 (q, C-21), 11.64 (q, C-18).
3.2.10. N-(3″-(3′,5′-Di-tert-butyl-4′-hydroxyphenyl)propyl)-3α,12α-dibenzyloxy-5β-cholane-24-amide (10)
Compound 1 (0.30 g, 0.52 mmol) and CDI (0.10 g, 0.62 mmol) were dissolved in CH2Cl2 (10 mL) and stirred at room temperature for 2 h. Then, 4-(3-aminopropyl)-2,6-di-tert-butylphenol (0.17 g, 0.62 mmol) was added and the reaction mixture was stirred at 30–35 °C for 8 h. The reaction course was monitored by TLC (CHCl3). Reaction mixture was diluted with CH2Cl2 and Et2O, washed sequentially with H2O and brine, and dried over anhydrous MgSO4. Solvent was evaporated to dryness; crude product (0.50 g, quantitative yield) was purified by flash column chromatography (SiO2 + top layer Al2O3, CH2Cl2 with gradient 0–5% AcOEt) to give a pure sample of compound 10 (0.35 g, 82%) as a white amorphous solid. Mp 65.0 °C [decomposition]. [] +48 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C55H79O4N1)+ m/z = 817.6004; found m/z = 817.5991. 1H-NMR (CDCl3, 600 MHz): δ = 7.39–7.24 (m, 10H, aromatic protons of Ph and Ph′), 6.97 (s, 2H, H-2^, H-6^), 5.40 (m, NH), 5.07 (s, OH), 4.62 (d, 1H, J = 11.5, H-26), 4.53 (m, CH2-25), 4.28 (d, 1H, J = 11.5, H-26′), 3.68 (s, 1H, H-12), 3.36 (m, 1H, H-3), 3.28 (m, 2H, CH2-1″), 2.56 (m, 2H, CH2-3″), 2.18 (m, 1H, H-23), 2.07–1.95 (m, 2H, H-23′, H-17), 1.91–0.86 (m, 51H, 1.44 (s, 18H, CH3-9^, 10^, 11^, 12^, 13^, 14^), 0.93 (s, 3H, CH3-19), 0.89 (d, 3H, J = 6.5, CH3-21)), 0.70 (s, 3H, CH3-18). 13C NMR (CDCl3, 150 MHz): δ = 173.33 (s, C-24), 151.76 (s, C-4^), 139.16 (s, C-1Ph *), 139.09 (s, C-1Ph′ *), 135.71 (d, C-3^, C-5^), 131.89 (s, C-1^), 128.10 (d, C-3Ph, C-5Ph #), 128.08 (d, C-3Ph′, C-5Ph′ #), 127.30 (d, C-2Ph, C-6Ph, C-2Ph′, C-6Ph′), 127.09 (d, C-4Ph §), 127.02 (d, C-4Ph′ §), 124.58 (d, C-2^, C-6^), 80.89 (d, C-12), 78.41 (d, C-3), 70.05 (t, C-26), 69.43 (t, C-25), 48.57 (d, C-14), 46.39 (s, C-13), 45.98 (d, C-17), 42.02 (d, C-5), 39.18 (t, C-1″), 35.92 (d, C-8), 35.21 (d, C-20), 35.16 (s, C-10‡), 34.37 (t, C-1‡), 34.11 (t, C-23), 33.62 (d, C-9), 33.33 (s, C-7^, C-8^ ∞), 33.15 (t, C-3″ ∞), 33.10 (t, C-4 ∞), 31.64 (t, C-22†), 31.54 (t, C-2″ †), 30.17 (q, C-9^, 10^, 11^, 12^, 13^, 14^), 27.48 (t, C-11¢), 27.21 (t, C-16¢), 27.88 (t, C-2¢), 25.88 (t, C-7), 23.56 (t, C-15), 23.21 (q, C-19), 22.88 (t, C-6), 17.41 (q, C-21), 12.60 (q, C-18).
3.2.11. N-(4′-Bromophenyl)-3α,12α-dibenzyloxy-5β-cholane-24-thioamide (11)
Compound 7 (0.10 g, 0.14 mmol) and Lawesson’s reagent (0.17 g, 0.41 mmol) in dry toluene (5 mL) were heated at 90–100 °C for 3 h under an argon atmosphere. Then, the reaction mixture was evaporated to dryness and chromatographed (SiO2, CH2Cl2) to give a pure sample of compound 11 (0.075 g, 72%) as an orange amorphous substance. Mp 58.0 °C [decomposition]. [] +35 (c 0.20 g/100 mL; CHCl3). HRMS: Calc. for (C44H56O2N79BrS)+ m/z = 741.3210; found m/z = 741.3207. Elemental analysis calculated for C44H56O2NBrS: C, 71.14; H, 7.60; Br, 10.76; N, 1.89; O, 4.31; S, 4.32; found C, 69.63; H, 7.42; N 1.99; S, 4.42. 1H-NMR (CDCl3, 400 MHz): δ = 8.63 (s, NH), 7.56–7.42 (m, 4H, H-2^, H-3^, H-5^, H-6^), 7.41–7.20 (m, 10H, aromatic protons Ph and Ph′), 4.62 (d, 1H, J = 11.5, H-26), 4.52 (s, CH2-25), 4.27 (d, 1H, J = 11.5, H-26′), 3.68 (s, 1H, H-12), 3.36 (m, 1H, H-3), 2.78 (m, 1H, H-23), 2.63 (m, 1H, H-23′), 2.12–0.84 (m, 30H, 2.05 (m, H-17), 0.94 (d, 3H, J = 6.5, CH3-21), 0.92 (s, 3H, CH3-19)), 0.70 (s, 3H, CH3-18). 13C NMR (CDCl3, 75 MHz): δ = 206.24 (s, C-24), 139.16 (s, C-1Ph *), 139.05 (s, C-1Ph′ *), 137.49 (s, C-1^), 131.76 (d, C-3^, C-5^), 128.14 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 127.37 (d, C-2Ph, C-6Ph #), 127.34 (d, C-2Ph′, C-6Ph′ #), 127.14 (d, C-4Ph §), 127.10 (d, C-4Ph′ §), 125.35 (d, C-2^, C-6^), 119.66 (s, C-4^), 80.88 (d, C-12), 78.45 (d, C-3), 70.07 (t, C-26), 69.48 (t, C-25), 48.66 (d, C-14), 46.47 (s, C-13), 45.94 (d, C-17), 45.47 (t, C-23), 42.02 (d, C-5), 35.93 (d, C-8), 35.70 (t, C-22), 35.16 (s, C-10; d, C-20), 34.38 (t, C-1), 33.66 (d, C-9), 33.12 (t, C-4), 27.59, 27.21, 27.12, 25.89 (t, C-7), 23.56 (t, C-15), 23.22 (q, C-19), 22.90 (t, C-6), 17.64 (q, C-21), 12.64 (q, C-18). Rotation around amide bond resulted in doubling of some signals in NMR 1H (NH, CH2-26, H-12, CH2-23). Rotamers ratio was about 85:15.
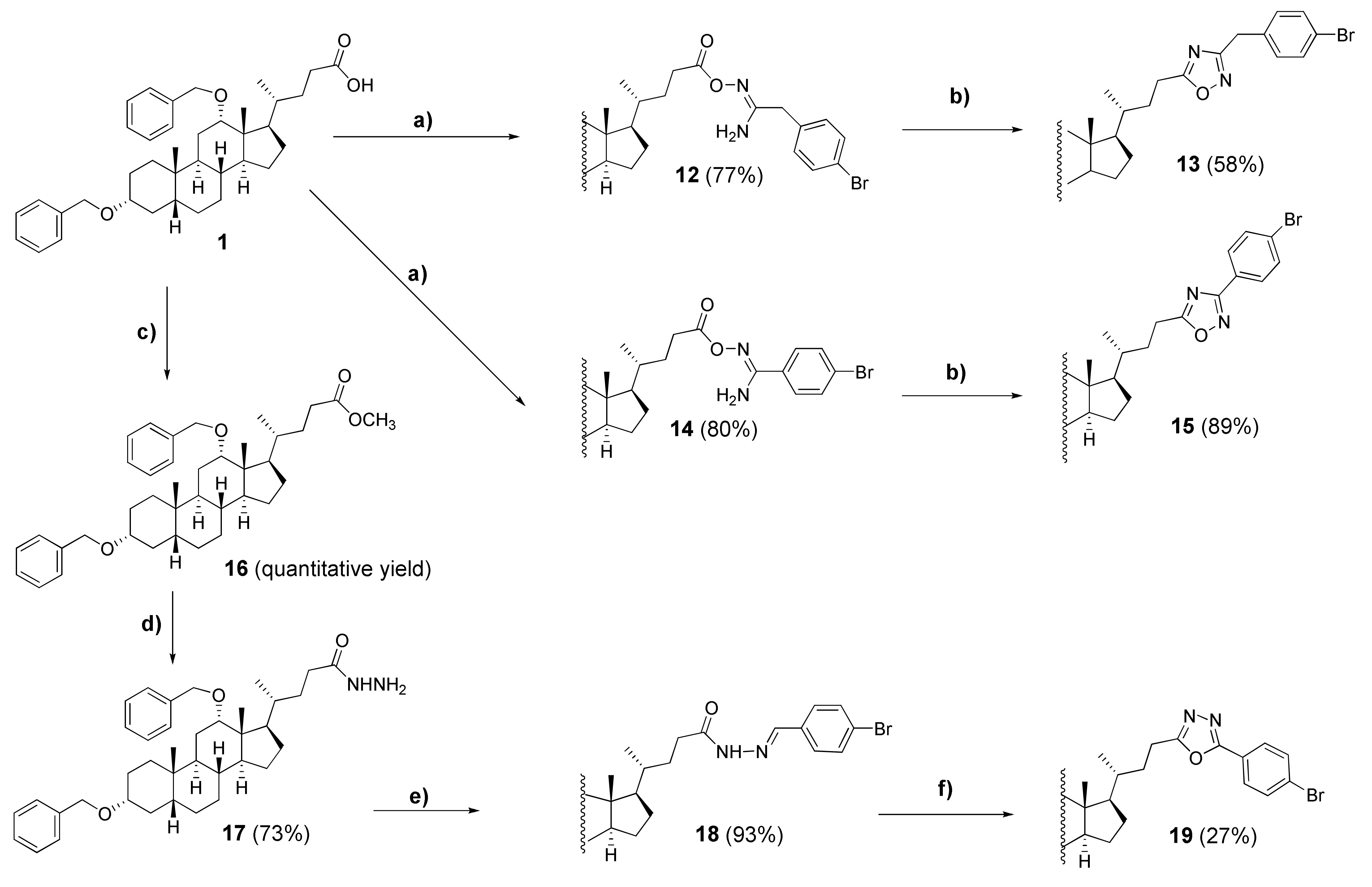
3.2.12. N′-(3α,12α-Dibenzyloxy-5β-cholane-24-oyl)-2-(4″-bromophenyl)acetimidamide (12)
Compound 1 (0.50 g, 0.87 mmol) and CDI (0.20 g, 1.21 mmol) were dissolved in dry CH2Cl2 (10 mL) and stirred at room temperature for 3 h. Then, 2-(4-bromophenyl)-N′-hydroxyacetimidamide (0.24 g, 1.21 mmol) was added and mixture was stirred at room temperature for 20 h rt, then refluxed for 3 h. The reaction course was monitored by TLC (CHCl3–MeOH, 60:1). The reaction mixture was evaporated to dryness and chromatographed (SiO2, CHC3) to give a pure sample of compound 12 (0.53 g, 77%) as a white amorphous solid. Mp 80.2–83.9 °C. [] +38 (c 0.20 g/100 mL; CHCl3). HRMS: m/z calcd. for (C46H59O4N279Br)+ 782.3653; found 764.3528; m/z calcd. for (C46H57O3N279Br)+ 764.3547 [M–H2O]+. 1H-NMR (CDCl3, 500 MHz): δ = 7.45–7.39 (m, 2H, H-3″, H-5″), 7.38–7.19 (m, 10H, aromatic protons of Ph and Ph′), 7.18–7.11 (m, 2H, H-2″, H-6″), 4.70 (br.s., NH2), 4.59 (d, 1H, J = 11.5, H-26), 4.51 (m, CH2-25), 4.27 (d, 1H, J = 11.5, H-26′), 3.66 (s, 1H, H-12), 3.46 (s, 2H, CH2-7″), 3.35 (m, 1H, H-3), 2.42 (m, 1H, H-23), 2.27 (m, 1H, H-23′), 2.03 (m, 1H, H-17), 1.91–0.83 (m, 29H, 0.91 (s, 3H, CH3-19), 0.90 (d, 3H, J = 6.0, CH3-21)), 0.69 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 171.94 (s, C-24(5′)), 156.04 (s, C-3′), 139.03 (s, C-1Ph, C-1Ph′), 133.90 (s, C-1″), 131.75 (d, C-3″, C-5″), 130.34 (d, C-2″, C-6″), 128.02 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 127.28 (d, C-2Ph, C-6Ph #), 127.21 (d, C-2Ph′, C-6Ph′ #), 127.01 (d, C-4Ph §), 126.97 (d, C-4Ph′ §), 121.20 (s, C-4″), 80.76 (d, C-12), 78.36 (d, C-3), 70.00 (t, C-26), 69.36 (t, C-25), 48.53 (d, C-14), 46.35 (s, C-13), 45.86 (d, C-17), 41.95 (d, C-5), 36.61 (t, C-7″), 35.85 (d, C-8), 35.09 (s, C-10‡), 34.98 (d, C-20), 34.28 (t, C-1‡), 33.57 (d, C-9), 33.05 (t, C-4), 30.80 (t, C-23†), 29.59 (t, C-22†), 27.33, 27.13, 27.04, 25.82 (t, C-7), 23.49 (t, C-15), 23.13 (q, C-19), 22.82 (t, C-6), 17.30 (q, C-21), 12.54 (q, C-18).
3.2.13. 5′-(24-Nor-3α,12α-dibenzyloxy-5β-cholan-23-yl)-3′-(methyl(4″-bromophenyl))-1′,2′,4′-oxadiazole (13)
Compound 12 (0.37 g, 0.47 mmol) and n-Bu4NF (1 M in THF; 0.5 mL, 0.47 mmol) in THF (8 mL) were refluxed for 3 h, then the reaction mixture was evaporated to dryness, redissolved in CH2Cl2–Et2O, washed with brine, and dried over anhydrous MgSO4. Solvent was evaporated under vacuum. Crude product (0.37 g, quantitative yield) was purified by flash column chromatography (SiO2, CHCl3) to give a pure sample of 13 (0.21 g, 58%) as a colorless amorphous mass. [] +38 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C46H57O3N279Br)+ m/z = 764.3547; found m/z = 673.3014; calc. for (C39H50O3N279Br)+ m/z = 673.3000 [M–PhCH2]+. 1H-NMR (CDCl3, 500 MHz): δ = 7.45–7.39 (m, 2H, H-3″, H-5″), 7.37–7.20 (m, 10H, aromatic protons of Ph and Ph′), 7.20–7.15 (m, 2H, H-2″, H-6″), 4.59 (d, 1H, J = 11.4, H-26), 4.52 (m, CH2-25), 4.26 (d, 1H, J = 11.4, H-26′), 3.97 (s, 2H, CH2-7″), 3.65 (s, 1H, H-12), 3.35 (m, 1H, H-3), 2.85 (m, 1H, H-23), 2.70 (m, 1H, H-23′), 2.04 (m, 1H, H-17), 1.94–0.81 (m, 29H, 0.91 (s, 3H, CH3-19), 0.90 (d, 3H, J = 6.6, CH3-21)), 0.65 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 180.68 (s, C-24 (5′)), 168.72 (s, C-3′), 139.21 (s, C-1Ph *), 139.10 (s, C-1Ph′ *), 134.49 (s, C-1″), 131.66 (d, C-3″, C-5″), 130.59 (d, C-2″, C-6″), 128.16 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 127.41 (d, C-2Ph, C-6Ph #), 127.35 (d, C-2Ph′, C-6Ph′ #), 127.14 (d, C-4Ph, C-4Ph′), 120.99 (s, C-4″), 80.79 (d, C-12), 78.48 (d, C-3), 70.12 (t, C-26), 69.51 (t, C-25), 48.62 (d, C-14), 46.52 (s, C-13), 45.96 (d, C-17), 42.12 (d, C-5), 36.01 (d, C-8), 35.25 (s, C-10‡), 35.14 (d, C-20), 34.44 (t, C-1‡), 33.69 (d, C-9), 33.19 (t, C-4), 32.58 (t, C-22†), 31.67 (t, C-7″ †), 27.45, 27.28, 27.19, 25.93 (t, C-7), 23.59 (t, C-15§), 23.37 (t, C-23§), 23.26 (q, C-19), 22.95, 17.29 (q, C-21), 12.60 (q, C-18).
3.2.14. N′-(3α,12α-Dibenzyloxy-5β-cholane-24-oyl)-4″-bromobenzimidamide (14)
Compound 1 (0.50 g, 0.87 mmol) and CDI (0.17 g, 1.2 mmol) were dissolved in dry CH2Cl2 (10 mL) and stirred at room temperature for 3 h. Then, 4-bromo-N′-hydroxybenzimidamide (0.23 g, 1.2 mmol) was added and the mixture was stirred at room temperature overnight. The reaction course was monitored by TLC (CHCl3). The reaction mixture was evaporated to dryness and chromatographed (SiO2, CHC3) to give a pure sample of compound 14 (0.54 g, 80%) as a white amorphous solid. Mp 144.5–147.4°C. [] +30 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C45H57O4N279Br)+ m/z = 768.3496; found m/z = 659.2835; calc. for (C38H48O3N279Br)+ m/z = 659.2843 [M–PhCH2–H2O]+. 1H-NMR (CDCl3, 500 MHz): δ = 7.57–7.48 (m, 2H, H-2″, H-3″, H-5″, H-6″), 7.39–7.21 (m, 10H, aromatic protons of Ph and Ph′), 5.06 (br.s., NH2), 4.60 (d, 1H, J = 11.5, H-26), 4.51 (m, CH2-25), 4.27 (d, 1H, J = 11.5, H-26′), 3.67 (s, 1H, H-12), 3.34 (m, 1H, H-3), 2.49 (m, 1H, H-23), 2.34 (m, 1H, H-23′), 2.03 (m, 1H, H-17), 1.92–0.83 (m, 29H, 0.91 (s, 3H, CH3-19), 0.90 (d, 3H, CH3-21)), 0.69 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 171.51 (s, C-24 (5′)), 155.08 (s, C-3′), 139.13 (s, C-1Ph *), 139.03 (s, C-1Ph′ *), 131.73 (d, C-3″, C-5″), 129.92 (s, C-1″), 128.14 (d, C-2″, C-6″ ∞), 128.12 (d, C-3Ph, C-5Ph ∞), 128.08 (d, C-3Ph′, C-5Ph′ ∞), 127.35 (d, C-2Ph, C-6Ph #), 127.33 (d, C-2Ph′, C-6Ph′ #), 127.14 (d, C-4Ph §), 127.06 (d, C-4Ph′ §), 125.21 (s, C-4″), 80.84 (d, C-12), 78.39 (d, C-3), 70.02 (t, C-26), 69.45 (t, C-25), 48.58 (d, C-14), 46.41 (s, C-13), 45.93 (d, C-17), 41.99 (d, C-5), 35.90 (d, C-8), 35.13 (s, C-10‡), 35.07 (d, C-20), 34.37 (t, C-1‡), 33.60 (d, C-9), 33.07 (t, C-4), 30.89 (t, C-23†), 29.68 (t, C-22†), 27.44, 27.20, 27.09, 25.88 (t, C-7), 23.57 (t, C-15), 23.23 (q, C-19), 22.84 (t, C-6), 17.34 (q, C-21), 12.64 (q, C-18).
3.2.15. 5′-(24-Nor-3α,12α-Dibenzyloxy-5β-cholan-23-yl)-3′-(4″-bromophenyl)-1′,2′,4′-oxadiazole (15)
Compound 14 (0.40 g, 0.52 mmol) and n-Bu4NF (1 M in THF; 0.25 mL, 0.25 mmol) in THF (5 mL) were refluxed for 1 h, then the reaction mixture was evaporated to dryness, dissolved in CH2Cl2–Et2O, washed with brine, and dried over anhydrous MgSO4. Solvent was evaporated under vacuum. Crude product (0.40 g, quantitative yield) was purified by flash column chromatography (SiO2, CH2Cl2) to give a pure sample of 15 (0.34 g, 89%) as a colorless amorphous solid. Mp 130.2–135.5 °C. [] +20 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C45H55O3N279Br)+ m/z = 750.3391; found m/z = 659.2839; calc. for (C38H48O3N279Br)+ m/z = 659.2843 [M–PhCH2]+. 1H-NMR (CDCl3, 400 MHz): δ = 7.93 (m, 2H, H-2″, H-6″), 7.60 (m, 2H, H-3″, H-5″), 7.40–7.20 (m, 10H, aromatic protons of Ph and Ph′), 4.61 (d, 1H, J = 11.5, H-26), 4.52 (m, CH2-25), 4.27 (d, 1H, J = 11.5, H-26′), 3.67 (s, 1H, H-12), 3.34 (m, 1H, H-3), 2.96 (m, 1H, H-23), 2.79 (m, 1H, H-23′), 2.08 (m, 1H, H-17), 2.03–0.86 (m, 29H, 0.94 (d, 3H, J = 6.5, CH3-21), 0.91 (s, 3H, CH3-19)), 0.69 (s, 3H, CH3-18). 13C NMR (CDCl3, 100 MHz): δ = 180.71 (s, C-24(5′)), 167.38 (s, C-3′), 139.10 (s, C-1Ph *), 139.08 (s, C-1Ph′ *), 131.96 (d, C-3″, C-5″), 128.77 (d, C-2″, C-6″), 128.17 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 127.37 (d, C-2Ph, C-6Ph, C-2Ph′, C-6Ph′), 127.15 (d, C-4Ph, C-4Ph′), 125.82 (s, C-1″ #), 125.43 (s, C-4″ #), 80.79 (d, C-12), 78.42 (d, C-3), 70.07 (t, C-26), 69.49 (t, C-25), 48.60 (d, C-14), 46.49 (s, C-13), 45.92 (d, C-17), 42.04 (d, C-5), 35.96 (d, C-8), 35.23 (d, C-20), 35.19 (s, C-10‡), 34.41(t, C-1‡), 33.63 (d, C-9), 33.12 (t, C-4), 32.64 (t, C-22), 27.50, 27.24, 27.14, 25.90 (t, C-7), 23.59 (t, C-15§), 23.35 (t, C-23§), 23.25 (q, C-19), 22.89, 17.28 (q, C-21), 12.65 (q, C-18).
3.2.16. Methyl 3α,12α-Dibenzyloxy-5β-cholan-24-oate (16)
Compound 1 (1.0 g, 1.75 mmol) and dimethylsulfate (0.45 mL, 4.81 mmol) were dissolved in acetone (50 mL); K2CO3 (0.89 g, 6.47 mmol) and KI (0.02 g, 0.12 mmol) were added to the solution. Reaction mixture was stirred overnight at room temperature. The reaction course was monitored by TLC (CHCl3). Then, acetone was evaporated under the vacuum. Reaction mixture was dissolved in CHCl3, washed with water and brine, and dried over anhydrous MgSO4. Solvent was evaporated under vacuum. Crude product (1.23 g) was purified by flash column chromatography (SiO2, CHCl3) to give pure 11 (1.1 g, quantitative yield) as a white amorphous solid. Mp 93.3–93.7 °C. [] +50 (c 0.20 g/100 mL; CHCl3). HRMS: Calc. for (C39H54O4)+ m/z = 586.4017; found m/z = 586.4022. 1H NMR (CDCl3, 500 MHz): δ = 7.40–7.22 (m, 10H, aromatic protons), 4.61 (d, 1H, J = 11.4, H-26), 4.53 (m, 2H, CH2-25), 4.29 (d, 1H, J = 11.4, H-26′), 3.68 (s, 1H, H-12), 3.66 (s, 3H, CH3-27), 3.36 (m, 1H, H-3), 2.37 (m, 1H, H-23), 2.21 (m, 1H, H-23′), 2.04 (m, 1H, H-17), 1.92–1.67 (m, 9H), 1.66–1.52 (m, 2H), 1.49–0.85 (m, 19H, 0.93 (s, 3H, CH3-19), 0.89 (d, 3H, J21,20 = 6.1, CH3-21)), 0.71 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 174.61 (s, C-24), 139.06 (s, C-1Ph, C-1Ph′), 128.08 (d, C-3Ph, C-5Ph *), 128.07 (d, C-3Ph′, C-5Ph′ *), 127.30 (d, C-2Ph, C-6Ph ∞), 127.28 (d, C-2Ph′, C-6Ph′ ∞), 127.07 (d, C-4Ph #), 127.02 (d, C-4Ph′ #), 80.79 (d, C-12), 78.36 (d, C-3), 70.03 (t, C-26), 69.39 (t, C-25), 51.26 (q, C-27), 48.51 (d, C-14), 46.37 (s, C-13), 45.97 (d, C-17), 41.98 (d, C-5), 35.89 (d, C-8), 35.13 (s, C-10§), 35.10 (d, C-20), 34.33 (t, C-1§), 33.56 (d, C-9), 33.04 (t, C-4), 30.71 (t, C-23‡), 30.75 (t, C-22‡), 27.40, (t, C-11†), 27.19 (t, C-16†), 27.07 (t, C-2†), 25.87 (t, C-7), 23.55 (t, C-15), 23.19 (q, C-19), 22.85 (t, C-6), 17.26 (q, C-21), 12.57 (q, C-18).
3.2.17. 3α,12α-Dibenzyloxy-5β-cholan-24-hydrazide (17)
Compound 16 (0.9 g, 1.54 mmol) and hydrazine hydrate (2.5 mL, 51.5 mmol) were dissolved in EtOH (25 mL), and solution was refluxed until full conversion was reached. The reaction course was monitored by TLC (CHCl3–MeOH; 25:0.5). Reaction mixture was concentrated under vacuum and poured into cold water. Precipitate was filtered, washed with water, and dried under vacuum. Crude product was purified by flash column chromatography (SiO2, CHCl3 with 0–2% MeOH) to give product 16 (0.66 g, 73%) and recrystallized from EtOH to give a pure sample of 17 as a white amorphous solid. Mp 179.0–180.0 °C (EtOH). [] +61 (c 0.20 g/100 mL; CHCl3). HRMS: m/z calcd. for (C38H54O3N2)+ 586.4129; found 586.4131. 1H NMR (CDCl3, 400 MHz): δ = 7.41–7.18 (m, 10H, aromatic protons), 5.24 (br.s., NH2), 4.58 (d, 1H, J = 11.4, H-26), 4.51 (m, 2H, CH2-25), 4.25 (d, 1H, J = 11.4, H-26′), 3.64 (s, 1H, H-12), 3.34 (m, 1H, H-3), 2.21 (m, 1H, H-23), 2.10–1.92 (m, 2H, H-23′, H-17), 1.92–0.80 (m, 29H, 0.90 (s, 3H, CH3-19), 0.86 (d, 3H, J21,20 = 5.8, CH3-21)), 0.67 (s, 3H, CH3-18). 13C NMR (CDCl3, 100 MHz): δ = 174.25 (s, C-24), 139.07 (s, C-1Ph, C-1Ph′), 128.10 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 127.33 (d, C-2Ph, C-6Ph *), 127.30 (d, C-2Ph′, C-6Ph′ *), 127.09 (d, C-4Ph, C-4Ph′), 80.83 (d, C-12), 78.37 (d, C-3), 70.07 (t, C-26), 69.38 (t, C-25), 48.52 (d, C-14), 46.38 (s, C-13), 46.03 (d, C-17), 41.99 (d, C-5), 35.89 (d, C-8), 35.20 (d, C-20), 35.13 (s, C-10§), 34.35 (t, C-1§), 33.58 (d, C-9), 33.06 (t, C-4), 31.22 (t, C-23‡), 30.98 (t, C-22‡), 27.47, (t, C-11†), 27.20 (t, C-16†), 27.08 (t, C-2†), 25.87 (t, C-7), 23.57 (t, C-15), 23.21 (q, C-19), 22.87 (t, C-6), 17.36 (q, C-21), 12.62 (q, C-18).
3.2.18. N’-(4″-Bromobenzylidene)-3α,12α-dibenzyloxy-5β-cholan-24-hydrazide (18)
Compound 17 (0.54 g, 0.93 mmol) and p-bromobenzaldehyde (0.17 g, 0.93 mmol) were dissolved in EtOH (10 mL), and the reaction mixture was refluxed for 3 h. The reaction course was monitored by TLC (CHCl3–MeOH; 30:0.5). Reaction mixture was evaporated to dryness and crude product was purified by flash column chromatography (SiO2, CHCl3) to give product 18 (0.65 g, 93%) as a white amorphous solid. Mp 147.7–150.0 °C (AcOEt). [] +48 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C45H57O3N279Br)+ m/z = 752.3547; found m/z = 661.2988; calc. for (C38H50O3N279Br)+ m/z [M–PhCH2]+ = 661.2999. 1H-NMR (CDCl3, 500 MHz): δ = 10.42 (s, NH&), 7.78 (s, H-5′ &), 7.56–7.45 (m, 4H, H-2″, H-3″, H-5″, H-6″), 7.41–7.18 (m, 10H, aromatic protons of Ph and Ph′), 4.62 (d, 1H, J = 11.4, H-26), 4.53 (m, CH2-25), 4.32 (d, 1H, J = 11.4, H-26′), 3.70 (s, 1H, H-12), 3.36 (m, 1H, H-3), 2.80 (m, 1H, H-23), 2.63 (m, 1H, H-23′), 2.11 (m, 1H, H-17), 1.96–0.88 (m, 29H, 0.99 (d, 3H, J = 6.0, CH3-21), 0.93 (s, 3H, CH3-19)), 0.72 (s, 3H, CH3-18). 13C NMR (CDCl3, 125 MHz): δ = 177.49 (s, C-24 (2′)), 142.28 (s, C-5′), 139.19 (s, C-1Ph *), 139.16 (s, C-1Ph′ *), 132.90 (s, C-1″), 131.78 (d, C-3″, C-5″), 128.31 (d, C-2″, C-6″), 128.12 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 127.33 (d, C-2Ph, C-6Ph #), 127.32 (d, C-2Ph′, C-6Ph′ #), 127.10 (d, C-4Ph ∞), 127.06 (d, C-4Ph′ ∞), 123.92 (s, C-4″), 80.90 (d, C-12), 78.48 (d, C-3), 70.15 (t, C-26), 69.46 (t, C-25), 48.62 (d, C-14), 46.50 (s, C-13), 46.15 (d, C-17), 42.09 (d, C-5), 36.00 (d, C-8), 35.52 (d, C-20), 35.23 (s, C-10‡), 34.42 (t, C-1‡), 33.69 (d, C-9), 33.16 (t, C-4), 31.03 (t, C-23§), 29.68 (t, C-22§), 27.54 (t, C-11‡), 27.27 (t, C-16‡), 27.17 (t, C-2‡), 25.93 (t, C-7), 23.61 (t, C-15), 23.24 (q, C-19), 22.97 (t, C-6), 17.61 (q, C-21), 12.67 (q, C-18).
3.2.19. 2′-(24-Nor-3α,12α-dibenzyloxy-5β-cholan-23-yl)-5′-(4″-bromophenyl)-1′,3′,4′-oxadiazole (19)
Compound 18 (0.57 g, 0.75 mmol) was dissolved in DMSO (10 mL) then I2 (0.29 g, 1.13 mmol) and potassium carbonate (0.39 g, 2.8 mmol) were added to the solution. Reaction mixture was heated at 70 °C for 3 h, then cooled to room temperature, diluted with Na2S2O3 (aq.), and stirred for 30 min. Reaction mixture was extracted with AcOEt–Et2O, washed with brine, and dried over anhydrous MgSO4. Solvent was evaporated to give crude product 19 (0.52 g, 93%). Crude product was chromatographed (SiO2, CH2Cl2 with 0–100% gradient CHCl3) to give pure compound 19 (0.15g, 27%). Mp 131.4 °C [decomposition]. [] +36 (c 0.10 g/100 mL; CHCl3). HRMS: Calc. for (C45H55O3N279Br)+ m/z = 750.3391; found m/z = 659.2838 and m/z = 750.3354; calc. for (C38H48O3N279Br)+ m/z = 659.2843. 1H-NMR (CDCl3, 300 MHz): δ = 7.88 (m, 2H, H-2″, H-6″), 7.62 (m, 2H, H-3″, H-5″), 7.44–7.18 (m, 10H, aromatic protons of Ph and Ph′), 4.61 (d, 1H, J = 11.5, H-26), 4.51 (m, CH2-25), 4.28 (d, 1H, J = 11.5, H-26′), 3.68 (s, 1H, H-12), 3.34 (m, 1H, H-3), 2.93 (m, 1H, H-23), 2.78 (m, 1H, H-23′), 2.19–0.82 (m, 30H; 2.09 (m, 1H, H-17), 0.96 (d, 3H, J = 6.3, CH3-21), 0.91 (s, 3H, CH3-19)), 0.69 (s, 3H, CH3-18). 13C NMR (CDCl3, 100 MHz): δ = 167.60 (s, C-5′), 163.83 (s, C-24 (2′)), 139.12 (s, C-1Ph, C-1Ph′), 132.22 (d, C-3″, C-5″), 128.17 (d, C-3Ph, C-5Ph, C-3Ph′, C-5Ph′), 128.05 (d, C-2″, C-6″), 127.38 (d, C-2Ph, C-6Ph, C-2Ph′, C-6Ph′), 127.16 (d, C-4Ph, C-4Ph′), 125.98 (s, C-1″), 122.91 (s, C-4″), 80.81 (d, C-12), 78.43 (d, C-3), 70.08 (t, C-26), 69.50 (t, C-25), 48.63 (d, C-14), 46.50 (s, C-13), 45.98 (d, C-17), 42.06 (d, C-5), 35.97 (d, C-8), 35.23 (d, C-20), 35.20 (s, C-10‡), 34.42 (t, C-1‡), 33.65 (d, C-9), 33.13 (t, C-4), 32.58 (t, C-22), 27.55 (t, C-11*), 27.24 (t, C-16*), 27.15 (t, C-2*), 25.91 (t, C-7), 23.60 (t, C-15), 23.25 (q, C-19), 22.90 (t, C-6), 22.21 (t, C-23), 17.31 (q, C-21), 12.65 (q, C-18).


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














