
Current Photoactive Molecules for Targeted Therapy of Triple-Negative Breast Cancer




Abstract
:1. Breast Cancer
1.1. Overview
1.2. Scope of the Review
1.3. Triple-Negative Breast Cancer
1.4. Triple-Negative Breast Cancer Therapeutics
2. Photothermal Therapy
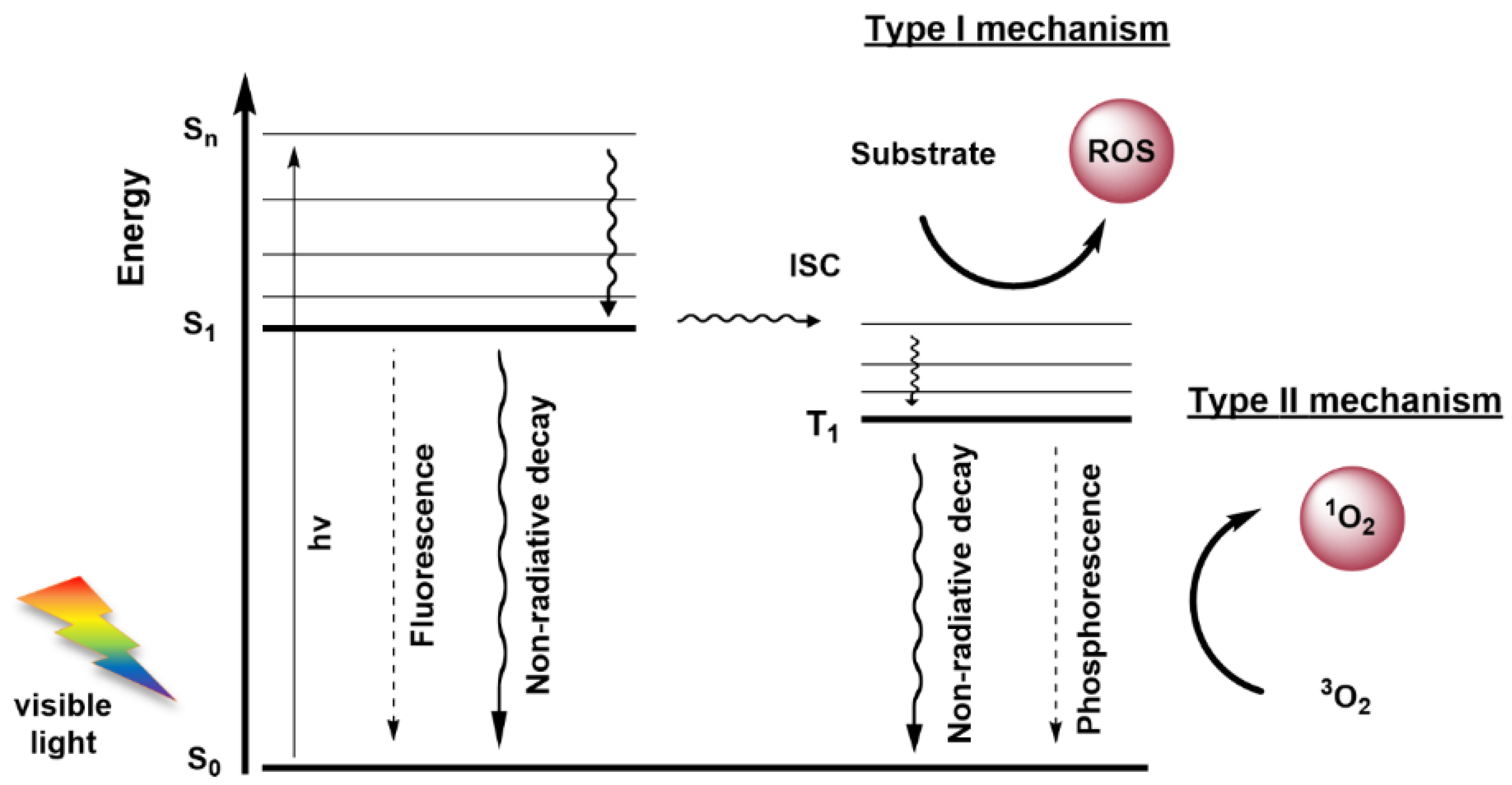
3. Photodynamic Therapy
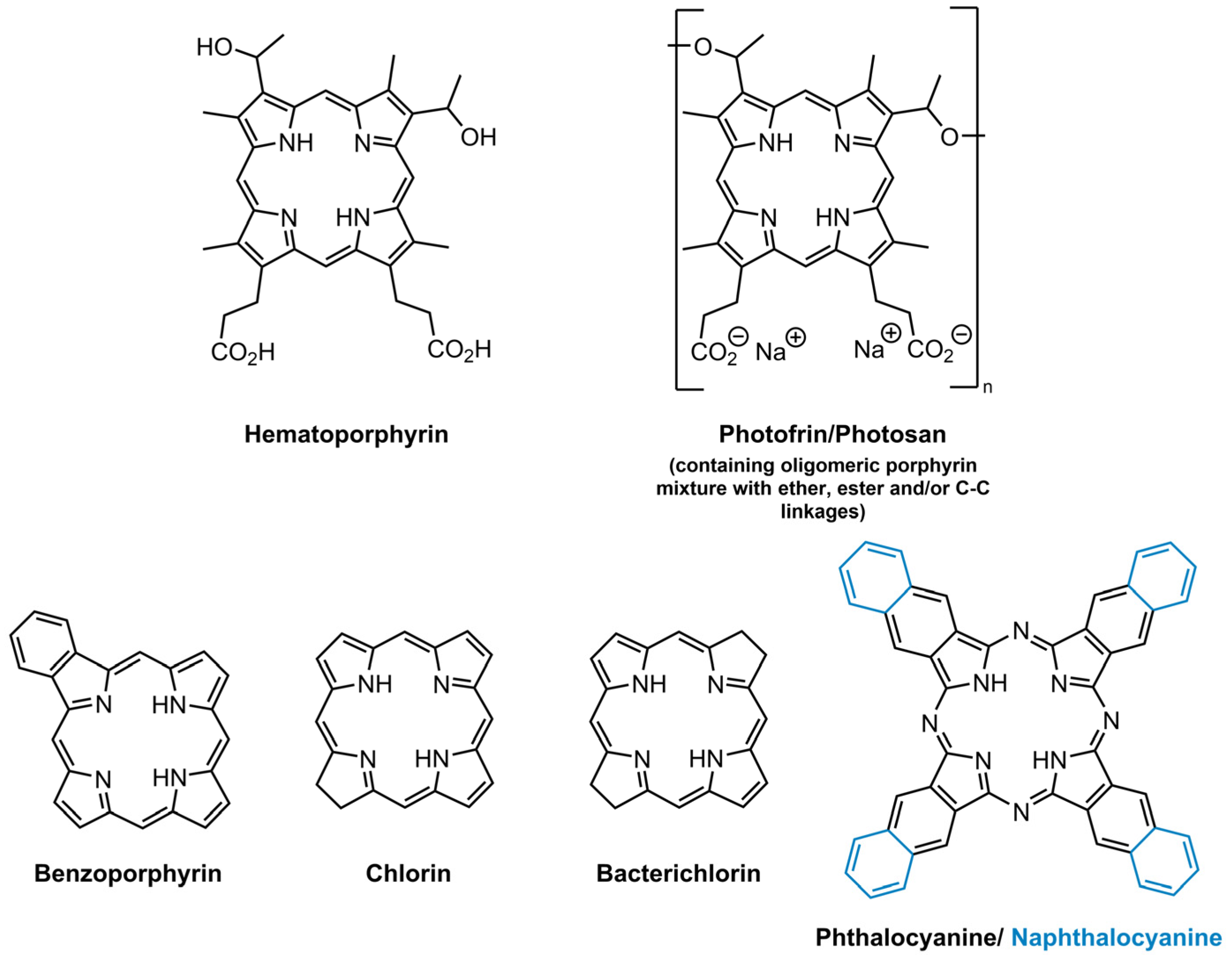
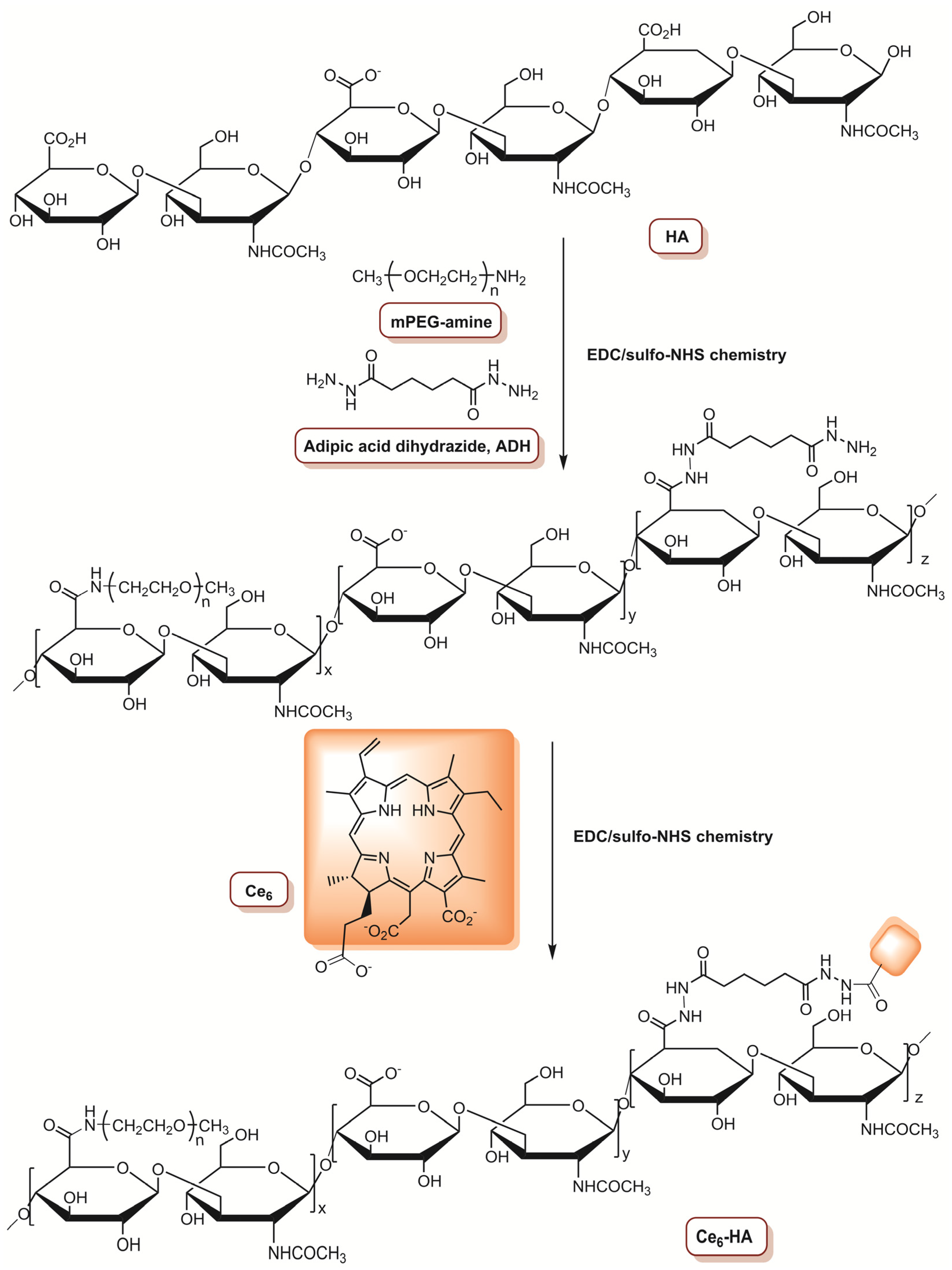
3.1. Photosensitizers
3.2. Anti-Cancer Mechanisms of Photodynamic Therapy
4. Targeted Therapy in Triple-Negative Breast Cancer
5. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Available online: https://gco.iarc.fr/today (accessed on 14 December 2021).
- Available online: https://gco.iarc.fr/tomorrow/ (accessed on 14 December 2021).
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Nagarajan, D.; McArdle, S.E.B. Immune landscape of breast cancers. Biomedicines 2018, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Pawar, A.; Prabhu, P. Nanosoldiers: A promising strategy to combat triple negative breast cancer. Biomed. Pharmacother. 2019, 110, 319–341. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Barry, M.; Gallagher, D.J.; Kell, M.; Sacchini, V. An overview of triple negative breast cancer for surgical oncologists. Surg. Oncol. 2015, 24, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Dolle, J.M.; Daling, J.R.; White, E.; Brinton, L.A.; Doody, D.R.; Porter, P.L.; Malone, K.E. Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol. Prev. Biomark. 2009, 18, 1157–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.S.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef]
- Pal, S.; Lüchtenborg, M.; Davies, E.A.; Jack, R.H. The treatment and survival of patients with triple negative breast cancer in a London population. J. Korean Phys. Soc. 2014, 3, 553. [Google Scholar] [CrossRef] [Green Version]
- Miller-Kleinhenz, J.M.; Bozeman, E.N.; Yang, L. Targeted nanoparticles for image-guided treatment of triple-negative breast cancer: Clinical significance and technological advances. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2015, 7, 797–816. [Google Scholar] [CrossRef] [Green Version]
- Dogan, B.E.; Gonzalez-Angulo, A.M.; Gilcrease, M.; Dryden, M.J.; Yang, W.T. Multimodality imaging of triple receptor-negative tumors with mammography, ultrasound, and MRI. Am. J. Roentgenol. 2010, 194, 1160–1166. [Google Scholar] [CrossRef]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2015, 8, 1913–1924. [Google Scholar] [CrossRef] [Green Version]
- Kalimutho, M.; Parsons, K.; Mittal, D.; López, J.A.; Srihari, S.; Khanna, K.K. Targeted Therapies for Triple-Negative Breast Cancer: Combating a Stubborn Disease. Trends Pharmacol. Sci. 2015, 36, 822–846. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.-P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]
- Emens, L.A. Immunotherapy in Triple-Negative Breast Cancer. Cancer J. 2021, 27, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Mediratta, K.; El-Sahli, S.; D’costa, V.; Wang, L. Current progresses and challenges of immunotherapy in triple-negative breast cancer. Cancers 2020, 12, 3529. [Google Scholar] [CrossRef]
- Segovia-Mendoza, M.; Romero-Garcia, S.; Lemini, C.; Prado-Garcia, H. Determining Factors in the Therapeutic Success of Checkpoint Immunotherapies against PD-L1 in Breast Cancer: A Focus on Epithelial-Mesenchymal Transition Activation. J. Immunol. Res. 2021, 2021, 6668573. [Google Scholar] [CrossRef]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pego, A.; Chan, A.; et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef]
- Litzenburger, B.C.; Creighton, C.J.; Tsimelzon, A.; Chan, B.T.; Susan, G.; Wang, T.; Carboni, J.M.; Gottardis, M.M.; Huang, F.; Jenny, C.; et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin. Cancer Res. 2011, 17, 2314–2327. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhou, Q.; Wang, H.; Zha, Y.; Zheng, P.; Yang, T.; Ma, D.; Qiu, L.; Xu, X.; Hu, Y.; et al. Mitochondria-targeted magnetic gold nanoheterostructure for multi-modal imaging guided photothermal and photodynamic therapy of triple-negative breast cancer. Chem. Eng. J. 2020, 403, 126364–126380. [Google Scholar] [CrossRef]
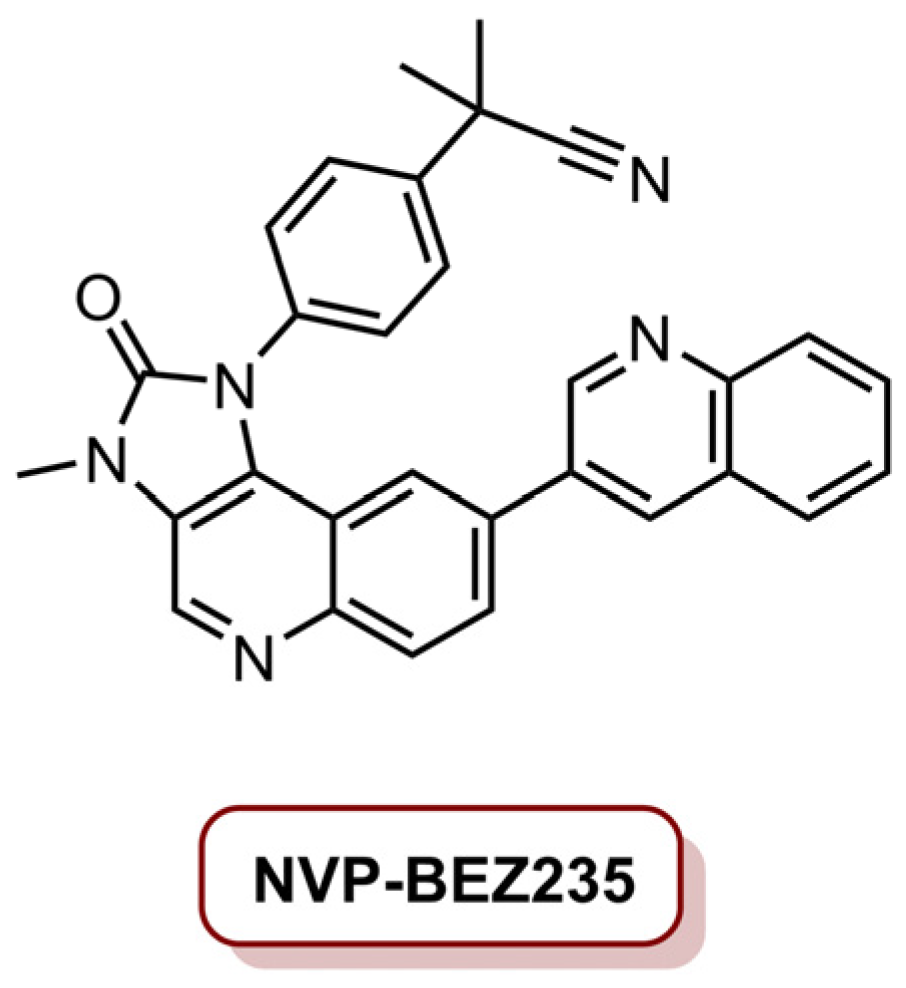
- Eltahan, A.S.; Liu, L.; Okeke, C.I.; Huang, M.; Han, L.; Chen, J.; Xue, X.; Bottini, M.; Guo, W.; Liang, X.J. NVP-BEZ235/Chlorin-e6 co-loaded nanoparticles ablate breast cancer by biochemical and photodynamic synergistic effects. Nano Res. 2018, 11, 4846–4858. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, S.; Zhang, G.; Sun, X.; Lee, S.T.; Liu, Z. Graphene in mice: Ultrahigh in vivo tumor uptake and efficient photothermal therapy. Nano Lett. 2010, 10, 3318–3323. [Google Scholar] [CrossRef]
- Liu, X.; Su, H.; Shi, W.; Liu, Y.; Sun, Y.; Ge, D. Functionalized poly(pyrrole-3-carboxylic acid) nanoneedles for dual-imaging guided PDT/PTT combination therapy. Biomaterials 2018, 167, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Wang, H.; He, B.; Zeng, L.; Tan, T.; Cao, H.; He, X.; Zhang, Z.; Guo, S.; Li, Y. Current Approaches of Photothermal Therapy in Treating Cancer Metastasis with Nanotherapeutics. Theranostics 2016, 6, 762–772. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Bao, X.; Cao, H.; Zhang, Z.; Yin, Q.; Gu, W. Tumor-Penetrating Nanotherapeutics Loading a Near-Infrared Probe Inhibit Growth and Metastasis of Breast Cancer. Adv. Funct. Mater. 2015, 25, 2831–2839. [Google Scholar] [CrossRef]
- Wang, D.; Xu, Z.; Yu, H.; Chen, X.; Feng, B.; Cui, Z.; Lin, B.; Yin, Q.; Zhang, Z.; Chen, C.; et al. Treatment of metastatic breast cancer by combination of chemotherapy and photothermal ablation using doxorubicin-loaded DNA wrapped gold nanorods. Biomaterials 2014, 35, 8374–8384. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Riedinger, A.; Li, H.; Fu, C.; Liu, H.; Li, L.; Liu, T.; Tan, L.; Barthel, M.J.; Pugliese, G.; et al. Plasmonic copper sulfide nanocrystals exhibiting near-infrared photothermal and photodynamic therapeutic effects. ACS Nano 2015, 9, 1788–1800. [Google Scholar] [CrossRef]
- Lin, M.; Guo, C.; Li, J.; Zhou, D.; Liu, K.; Zhang, X.; Xu, T.; Zhang, H.; Wang, L.; Yang, B. Polypyrrole-coated chainlike gold nanoparticle architectures with the 808 nm photothermal transduction efficiency up to 70%. ACS Appl. Mater. Interfaces 2014, 6, 5860–5868. [Google Scholar] [CrossRef]
- Tang, S.; Chen, M.; Zheng, N. Sub-10-nm Pd nanosheets with renal clearance for efficient near-infrared photothermal cancer therapy. Small 2014, 10, 3139–3144. [Google Scholar] [CrossRef]
- Robinson, J.T.; Tabakman, S.M.; Liang, Y.; Wang, H.; Sanchez Casalongue, H.; Vinh, D.; Dai, H. Ultrasmall reduced graphene oxide with high near-infrared absorbance for photothermal therapy. J. Am. Chem. Soc. 2011, 133, 6825–6831. [Google Scholar] [CrossRef]
- Liang, C.; Diao, S.; Wang, C.; Gong, H.; Liu, T.; Hong, G.; Shi, X.; Dai, H.; Liu, Z. Tumor metastasis inhibition by imaging-guided photothermal therapy with single-walled carbon nanotubes. Adv. Mater. 2014, 26, 5646–5652. [Google Scholar] [CrossRef]
- Ma, Y.; Liang, X.; Tong, S.; Bao, G.; Ren, Q.; Dai, Z. Gold nanoshell nanomicelles for potential magnetic resonance imaging, light-triggered drug release, and photothermal therapy. Adv. Funct. Mater. 2013, 23, 815–822. [Google Scholar] [CrossRef]
- Hu, S.H.; Fang, R.H.; Chen, Y.W.; Liao, B.J.; Chen, I.W.; Chen, S.Y. Photoresponsive protein-graphene-protein hybrid capsules with dual targeted heat-triggered drug delivery approach for enhanced tumor therapy. Adv. Funct. Mater. 2014, 24, 4144–4155. [Google Scholar] [CrossRef]
- Mesquita, M.Q.; Dias, C.J.; Neves, M.G.P.M.S.; Almeida, A.; Faustino, M.A.F. Revisiting current photoactive materials for antimicrobial photodynamic therapy. Molecules 2018, 23, 2424. [Google Scholar] [CrossRef] [Green Version]
- Mesquita, M.Q.; Dias, C.J.; Gamelas, S.; Fardilha, M.; Neves, M.G.P.M.S.; Faustino, M.A.F. An insight on the role of photosensitizer nanocarriers for Photodynamic Therapy. Ann. Braz. Acad. Sci. 2018, 90, 1101–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamse, H.; Hamblin, M. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Rumie Vittar, N.B.; Rivarola, V.A. Breast cancer as photodynamic therapy target: Enhanced therapeutic efficiency by overview of tumor complexity. World J. Clin. Oncol. 2014, 5, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, A.E.; Boschi, F. Photodynamic therapy using Cerenkov and radioluminescence light. Front. Phys. 2021, 9, 80. [Google Scholar] [CrossRef]
- Hartl, B.A.; Hirschberg, H.; Marcu, L.; Cherry, S.R. Activating photodynamic therapy in vitro with cerenkov radiation generated from yttrium-90. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Pinto da Silva, L.; Magalhães, C.M.; Núñez-Montenegro, A.; Ferreira, P.J.O.; Duarte, D.; Rodríguez-Borges, J.E.; Vale, N.; Esteves da Silva, J.C.G. Study of the combination of self-activating photodynamic therapy and chemotherapy for cancer treatment. Biomolecules 2019, 9, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, L.P.; Nunez-Montenegro, A.; Magalhaes, C.M.; Ferreira, P.J.O.; Duarte, D.; Gonzalez-Berdullas, P.; Rodriguez-Borges, J.E.; Vale, N.; da Silva, J.C.G.E. Single-molecule chemiluminescent photosensitizer for a self-activating and tumor-selective photodynamic therapy of cancer. Eur. J. Med. Chem. 2019, 183, 111683. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zhou, Z.; Pratx, G.; Chen, X.; Chen, H. Nanoscintillator-mediated X-ray induced photodynamic therapy for deep-seated tumors: From concept to biomedical applications. Theranostics 2020, 10, 1296. [Google Scholar] [CrossRef]
- Larue, L.; Mihoub, A.B.; Youssef, Z.; Colombeau, L.; Acherar, S.; André, J.-C.; Arnoux, P.; Baros, F.; Vermandel, M.; Frochot, C. Using X-rays in photodynamic therapy: An overview. Photochem. Photobiol. Sci. 2018, 17, 1612–1650. [Google Scholar] [CrossRef]
- Banerjee, S.M.; Macrobert, A.J.; Mosse, C.A.; Periera, B.; Bown, S.G.; Keshtgar, M.R.S. Photodynamic therapy: Inception to application in breast cancer. Breast 2017, 31, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Dąbrowski, J.M.; Arnaut, L.G. Photodynamic therapy (PDT) of cancer: From local to systemic treatment. Photochem. Photobiol. Sci. 2015, 14, 1765–1780. [Google Scholar] [CrossRef]
- Donohoe, C.; Senge, M.O.; Arnaut, L.G.; Gomes-da-Silva, L.C. Cell death in photodynamic therapy: From oxidative stress to anti-tumor immunity. Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 188308–188324. [Google Scholar] [CrossRef] [PubMed]
- Direito, I.; Fardilha, M.; Helguero, L.A. Contribution of the unfolded protein response to breast and prostate tissue homeostasis and its significance to cancer endocrine response. Carcinogenesis 2019, 40, 203–215. [Google Scholar] [CrossRef]
- Li, W.; Kong, A.N. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol. Carcinog. 2009, 48, 91–104. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Kessel, D.; Oleinick, N.L. Cell Death Pathways Associated with Photodynamic Therapy: An Update. Photochem. Photobiol. 2018, 94, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, V.; Abbas, A.; Aster, J. Robbins Basic Pathology, 10th ed.; Elsevier: Philadelphia, PA, USA, 2017. [Google Scholar]
- Falk-Mahapatra, R.; Gollnick, S.O. Photodynamic Therapy and Immunity: An Update. Photochem. Photobiol. 2020, 96, 550–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapozzi, V.; D’Este, F.; Xodo, L.E. Molecular pathways in cancer response to photodynamic therapy. J. Porphyr. Phthalocyanines 2019, 23, 410–418. [Google Scholar] [CrossRef]
- Kessel, D. Apoptosis, Paraptosis and Autophagy: Death and Survival Pathways Associated with Photodynamic Therapy. Photochem. Photobiol. 2019, 95, 119–125. [Google Scholar] [CrossRef] [Green Version]
- Ram, B.M.; Ramakrishna, G. Endoplasmic reticulum vacuolation and unfolded protein response leading to paraptosis like cell death in cyclosporine A treated cancer cervix cells is mediated by cyclophilin B inhibition. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2497–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessel, D.; Reiners, J.J.J. Effects of Combined Lysosomal and Mitochondrial Photodamage in a Non-small-Cell Lung Cancer Cell Line: The Role of Paraptosis. Photochem. Photobiol. 2017, 93, 1502–1508. [Google Scholar] [CrossRef]
- Pierroz, V.; Rubbiani, R.; Gentili, C.; Patra, M.; Mari, C.; Gasser, G.; Ferrari, S. Dual mode of cell death upon the photo-irradiation of a RuII polypyridyl complex in interphase or mitosis. Chem. Sci. 2016, 7, 6115–6124. [Google Scholar] [CrossRef] [Green Version]
- Gomes-da-Silva, L.C.; Zhao, L.; Bezu, L.; Zhou, H.; Sauvat, A.; Liu, P.; Durand, S.; Leduc, M.; Souquere, S.; Loos, F.; et al. Photodynamic therapy with redaporfin targets the endoplasmic reticulum and Golgi apparatus. EMBO J. 2018, 37, e98354. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Enlightening the impact of immunogenic cell death in photodynamic cancer therapy. EMBO J. 2012, 31, 1055–1057. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Xu, Y.; Fu, P.; Chen, M.; Sun, S.; Zhao, R.; Wang, J.; Liang, X.; Wang, S. Ultrasound-targeted photodynamic and gene dual therapy for effectively inhibiting triple negative breast cancer by cationic porphyrin lipid microbubbles loaded with HIF1α-siRNA. Nanoscale 2018, 10, 19945–19956. [Google Scholar] [CrossRef]
- Jadia, R.; Kydd, J.; Rai, P. Remotely Phototriggered, Transferrin-Targeted Polymeric Nanoparticles for the Treatment of Breast Cancer. Photochem. Photobiol. 2018, 94, 765–774. [Google Scholar] [CrossRef]
- Potocny, A.M.; Riley, R.S.; 0′Sullivan, R.K.; Day, E.S.; Rosenthal, J. Photochemotherapeutic Properties of a Linear Tetrapyrrole Palladium(II) Complex displaying an Exceptionally High Phototoxicity Index. Inorg. Chem. 2018, 57, 10608–10615. [Google Scholar] [CrossRef] [PubMed]
- Juneja, R.; Lyles, Z.; Vadarevu, H.; Afonin, K.A.; Vivero-escoto, J.L. Multimodal Polysilsesquioxane Nanoparticles for Combinatorial Therapy and Gene Delivery in Triple-Negative Breast Cancer. ACS Appl. Mater. Interfaces 2019, 11, 12308–12320. [Google Scholar] [CrossRef]
- Sakamaki, Y.; Ozdemir, J.; Perez, A.D.; Heidrick, Z.; Watson, O.; Tsuji, M.; Salmon, C.; Batta-mpouma, J.; Azzun, A.; Lomonte, V.; et al. Maltotriose Conjugated Metal—Organic Frameworks for Selective Targeting and Photodynamic Therapy of Triple Negative Breast Cancer Cells and Tumor Associated Macrophages. Adv. Ther. 2020, 3, 2000029. [Google Scholar] [CrossRef]
- Huang, P.; Zhang, B.; Yuan, Q.; Zhang, X.; Leung, W.; Xu, C. Photodynamic treatment with purpurin 18 effectively inhibits triple negative breast cancer by inducing cell apoptosis. Lasers Med. Sci. 2021, 36, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Lyles, Z.K.; Tarannum, M.; Mena, C.; Inada, N.M.; Bagnato, V.S.; Vivero-escoto, J.L. Biodegradable Silica-Based Nanoparticles with Improved and Safe Delivery of Protoporphyrin IX for the In Vivo Photodynamic Therapy of Breast Cancer. Adv. Ther. 2020, 3, 2000022. [Google Scholar] [CrossRef]
- Chung, C.; Lu, K.; Lee, W.; Hsu, W.; Lee, W.; Dai, J.; Shueng, P.; Lin, C.; Mi, F. Fucoidan-based, tumor-activated nanoplatform for overcoming hypoxia and enhancing photodynamic therapy and antitumor immunity. Biomaterials 2020, 257, 120227. [Google Scholar] [CrossRef]
- Aru, B.; Gunay, A.; Şenkuytu, E.; Demirel, G.Y.; Gürek, A.G.; Atilla, D. A Translational Study of a Silicon Phthalocyanine Substituted with a Histone Deacetylase Inhibitor for Photodynamic Therapy. ACS Omega 2020, 5, 25854–25867. [Google Scholar] [CrossRef]
- Li, X.; Jeon, Y.; Kwon, N.; Park, J.; Guo, T.; Kim, H.; Huang, J.; Lee, D.; Yoon, J. In Vivo-assembled phthalocyanine/albumin supramolecular complexes combined with a hypoxia-activated prodrug for enhanced photodynamic immunotherapy of cancer. Biomaterials 2021, 266, 120430. [Google Scholar] [CrossRef]
- Choi, J.; Kim, H.; Choi, Y. Theranostic nanoparticles for enzyme-activatable fluorescence imaging and photodynamic/chemo dual therapy of triple-negative breast cancer. Quant. Imaging Med. Surg. 2015, 5, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Won, Y.; Goh, S.H.; Choi, Y. A redox-responsive theranostic agent for target-specific fluorescence imaging and photodynamic therapy of EGFR-overexpressing triple-negative breast cancers. J. Mater. Chem. B 2016, 4, 6787–6790. [Google Scholar] [CrossRef]
- Feng, B.; Zhou, F.; Xu, Z.; Wang, T.; Wang, D.; Liu, J.; Fu, Y.; Yin, Q.; Zhang, Z.; Yu, H.; et al. Versatile Prodrug Nanoparticles for Acid-Triggered Precise Imaging and Organelle-Specific Combination Cancer Therapy. Adv. Funct. Mater. 2016, 26, 7431–7442. [Google Scholar] [CrossRef]
- Zhou, F.; Feng, B.; Wang, T.; Wang, D.; Meng, Q.; Zeng, J.; Zhang, Z.; Wang, S.; Yu, H.; Li, Y. Programmed Multiresponsive Vesicles for Enhanced Tumor Penetration and Combination Therapy of Triple-Negative Breast Cancer. Adv. Funct. Mater. 2017, 27, 1606530. [Google Scholar] [CrossRef]
- Zhou, F.; Feng, B.; Wang, T.; Wang, D.; Cui, Z.; Wang, S.; Ding, C.; Zhang, Z.; Liu, J.; Yu, H.; et al. Theranostic Prodrug Vesicles for Reactive Oxygen Species-Triggered Ultrafast Drug Release and Local-Regional Therapy of Metastatic Triple-Negative Breast Cancer. Adv. Funct. Mater. 2017, 27, 1703674. [Google Scholar] [CrossRef]
- Bharathiraja, S.; Manivasagan, P.; Moorthy, M.S.; Bui, N.Q.; Lee, K.D.; Oh, J. Chlorin e6 conjugated copper sulfide nanoparticles for photodynamic combined photothermal therapy. Photodiagnosis Photodyn. Ther. 2017, 19, 128–134. [Google Scholar] [CrossRef]
- Meng, Q.; Meng, J.; Ran, W.; Wang, J.; Zhai, Y.; Zhang, P.; Li, Y. Light-Activated Core-Shell Nanoparticles for Spatiotemporally Specific Treatment of Metastatic Triple-Negative Breast Cancer. ACS Nano 2018, 12, 2789–2802. [Google Scholar] [CrossRef]
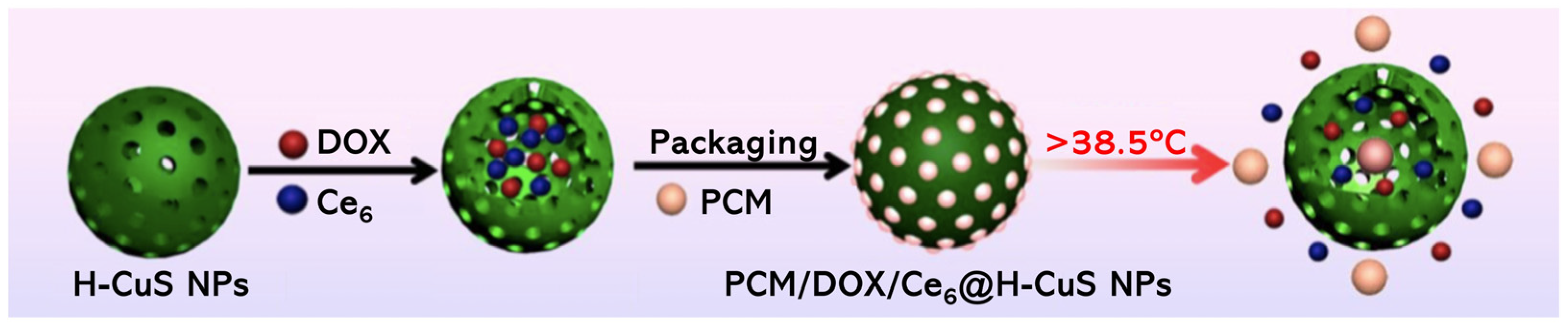
- Li, Q.; Sun, L.; Hou, M.; Chen, Q.; Ruihao, Y.; Zhang, L.; Xu, Z.; Kang, Y.; Xue, P. Phase-Change Material Packaged within Hollow Copper Sulfide Nanoparticles Carrying Doxorubicin and Chlorin e6 for Fluorescence-Guided Trimodal Therapy of Cancer. ACS Appl. Mater. Interfaces 2018, 11, 417–429. [Google Scholar] [CrossRef]
- Zhou, T.-J.; Xing, L.; Fan, Y.-T.; Cui, P.-F.; Jiang, H.-L. Inhibition of breast cancer proliferation and metastasis by strengthening host immunity with a prolonged oxygen-generating phototherapy hydrogel. J. Control. Release 2019, 309, 82–93. [Google Scholar] [CrossRef]
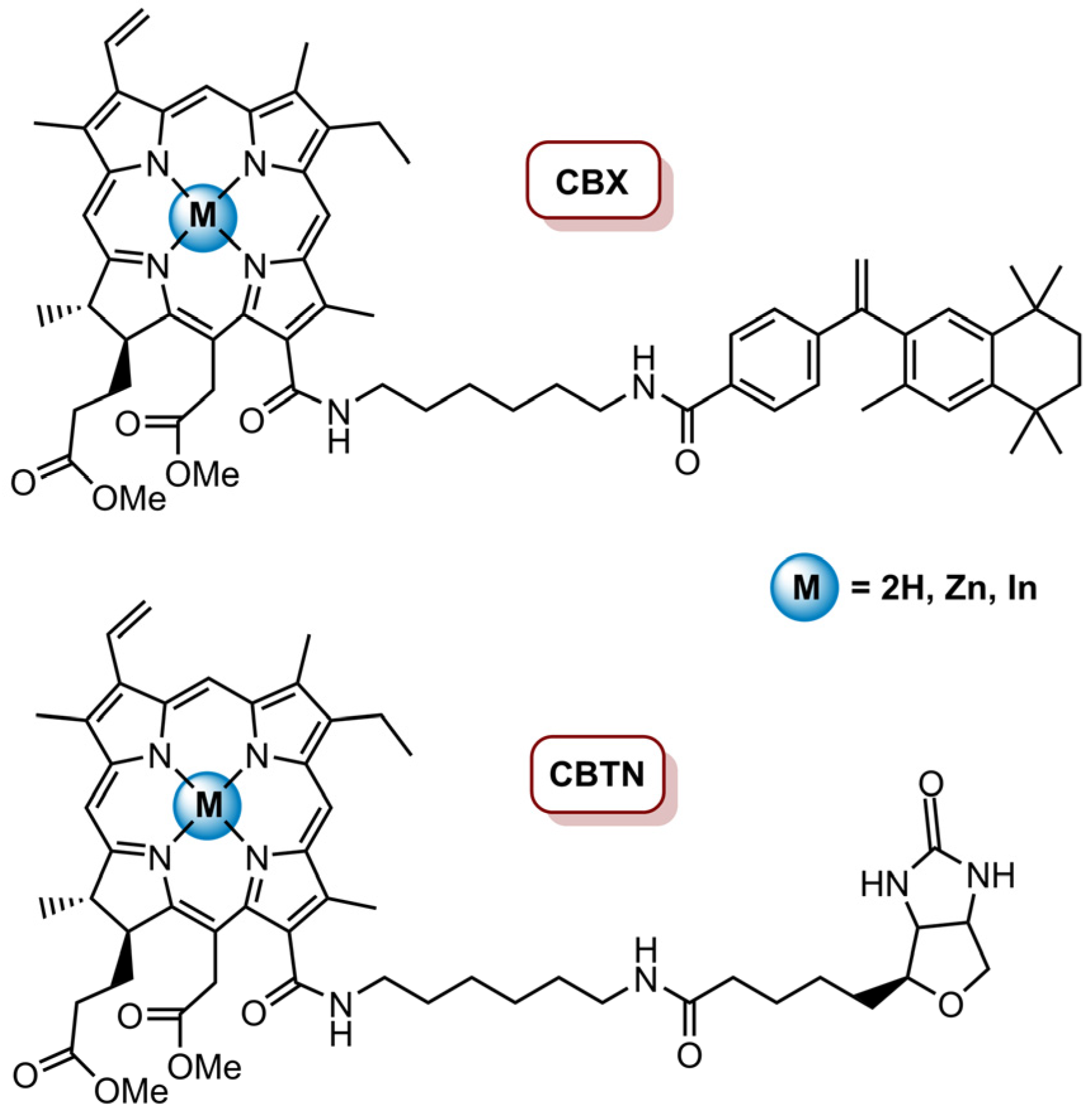
- Isaac-Lam, M.F.; Mee, A.D. Photodynamic Activity of Vitamin-Chlorin Conjugates at Nanomolar Concentrations against Triple-Negative Breast Cancer Cells. ACS Omega 2019, 4, 2907–2920. [Google Scholar] [CrossRef]
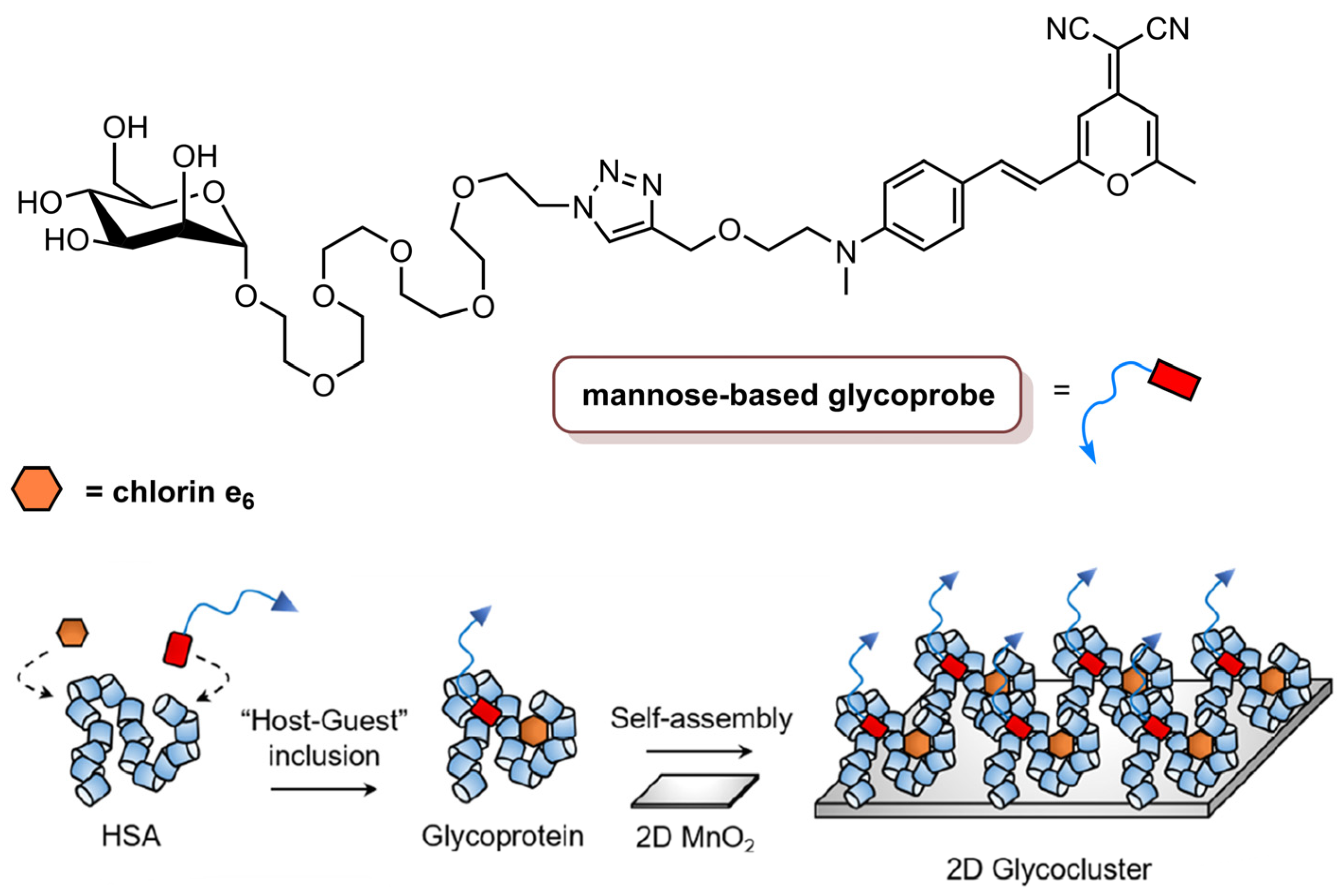
- Hu, X.L.; Cai, Q.; Gao, J.; Field, R.A.; Chen, G.R.; Jia, N.; Zang, Y.; Li, J.; He, X.P. Self-Assembled 2D Glycoclusters for the Targeted Delivery of Theranostic Agents to Triple-Negative Breast Cancer Cells. ACS Appl. Mater. Interfaces 2019, 11, 22181–22187. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Z.; Ren, J.; Ba, L.; He, W.; Wong, C.Y. Multifunctional oxygen-enriching nano-theranostics for cancer-specific magnetic resonance imaging and enhanced photodynamic/photothermal therapy. Nano Res. 2020, 13, 1389–1398. [Google Scholar] [CrossRef]
- Li, Z.; Yang, F.; Wu, D.; Liu, Y.; Gao, Y.; Lian, H.; Zhang, H.; Yin, Z.; Wu, A.; Zeng, L. Ce6-Conjugated and polydopamine-coated gold nanostars with enhanced photoacoustic imaging and photothermal/photodynamic therapy to inhibit lung metastasis of breast cancer. Nanoscale 2020, 12, 22173–22184. [Google Scholar] [CrossRef] [PubMed]
- Zduniak, K.; Gdesz-Birula, K.; Woźniak, M.; Duś-Szachniewicz, K.; Ziółkowski, P. The Assessment of the Combined Treatment of 5-ALA Mediated Photodynamic Therapy and Thalidomide on 4T1 Breast Carcinoma and 2H11 Endothelial Cell Line. Molecules 2020, 25, 5184. [Google Scholar] [CrossRef] [PubMed]
- Guney Eskiler, G.; Deveci Ozkan, A.; Sozen Kucukkara, E.; Kamanlı, A.F.; Gunoğlu, B.; Yıldız, M.Z. Optimization of 5-aminolevulinic acid-based photodynamic therapy protocol for breast cancer cells. Photodiagnosis Photodyn. Ther. 2020, 31, 101854. [Google Scholar] [CrossRef]
- Palasuberniam, P.; Kraus, D.; Mansi, M.; Howley, R.; Braun, A.; Myers, K.A.; Chen, B. Small molecule kinase inhibitors enhance aminolevulinic acid-mediated protoporphyrin IX fluorescence and PDT response in triple negative breast cancer cell lines. J. Biomed. Opt. 2021, 26, 098002. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Hardy, C.W.; Yu, D.S.; Fernandez, B.; Zhang, H. Indocyanine green loaded liposome nanocarriers for photodynamic therapy using human triple negative breast cancer cells. Photodiagnosis Photodyn. Ther. 2014, 11, 193–203. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Moshkelani, D.; Zhang, H. Thermosensitive Liposome Formulated Indocyanine Green for Near-Infrared Triggered Photodynamic Therapy: In Vivo Evaluation for Triple-Negative Breast Cancer. Pharm. Res. 2015, 32, 1604–1614. [Google Scholar] [CrossRef]
- Wu, C.; Tian, Y.; Zhang, Y.; Xu, J.; Wang, Y.; Guan, X. Acid-Triggered Charge-Convertible Graphene-Based All-in-One Nanocomplex for Enhanced Genetic Phototherapy of Triple-Negative Breast Cancer. Adv. Healthc. Mater. 2019, 9, e1901187. [Google Scholar] [CrossRef]
- Higbee-dempsey, E.; Amirshaghaghi, A.; Case, M.J.; Miller, J.; Busch, T.M.; Tsourkas, A. Indocyanine Green—Coated Gold Nanoclusters for Photoacoustic Imaging and Photothermal Therapy. Adv. Ther. 2019, 2, 1900088. [Google Scholar] [CrossRef]
- Liu, L.; He, H.; Luo, Z.; Zhou, H.; Liang, R.; Pan, H. In Situ Photocatalyzed Oxygen Generation with Photosynthetic Bacteria to Enable Robust Immunogenic Photodynamic Therapy in Triple-Negative Breast Cancer. Adv. Funct. Mater. 2020, 30, 1910176. [Google Scholar] [CrossRef]
- Pucelik, B.; Sułek, A.; Da, J.M. Bacteriochlorins and their metal complexes as NIR-absorbing photosensitizers: Properties, mechanisms, and applications. Coord. Chem. Rev. 2020, 416, 213340. [Google Scholar] [CrossRef]
- Medina, M.A.; Oza, G.; Sharma, A.; Arriaga, L.G.; Hern, M.; Rotello, V.M.; Ramirez, J.T. Triple-Negative Breast Cancer: A Review of Conventional and Advanced Therapeutic Strategies. Int. J. Environ. Res. Public Health 2020, 17, 2078. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Sun, Z.; Ren, Y.; Chen, X.; Zhang, W.; Zhu, X.; Mao, Z.; Shen, J.; Nie, S. Advances in nanomaterials for use in photothermal and photodynamic therapeutics. Mol. Med. Rep. 2019, 20, 5–15. [Google Scholar] [CrossRef] [Green Version]
- del Alcazar, C.R.G.; Gillam, M.C.; Mukherjee, B.; Tomimatsu, N.; Gao, X.; Yan, J.; Xie, X.-J.; Bachoo, R.; Li, L.; Habib, A.A.; et al. Inhibition of DNA Double-Strand Break Repair by the Dual PI3K/mTOR Inhibitor NVP-BEZ235 as a Strategy for Radiosensitization of Glioblastoma. Clin. Cancer Res. 2014, 20, 1235–1248. [Google Scholar] [CrossRef] [Green Version]
- Hyun, D.; Lu, P.; Choi, S.-I.; Jeong, U.; Xia, Y. Microscale Polymer Bottles Corked with a Phase-Change Material for Temperature-Controlled Release. Angew. Chem. Int. Ed. Engl. 2013, 52, 10468–10471. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Zhang, Y.; Xia, Y. A temperature-sensitive drug release system based on phase-change materials. Angew. Chem. Int. Ed. 2010, 49, 7904–7908. [Google Scholar] [CrossRef] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
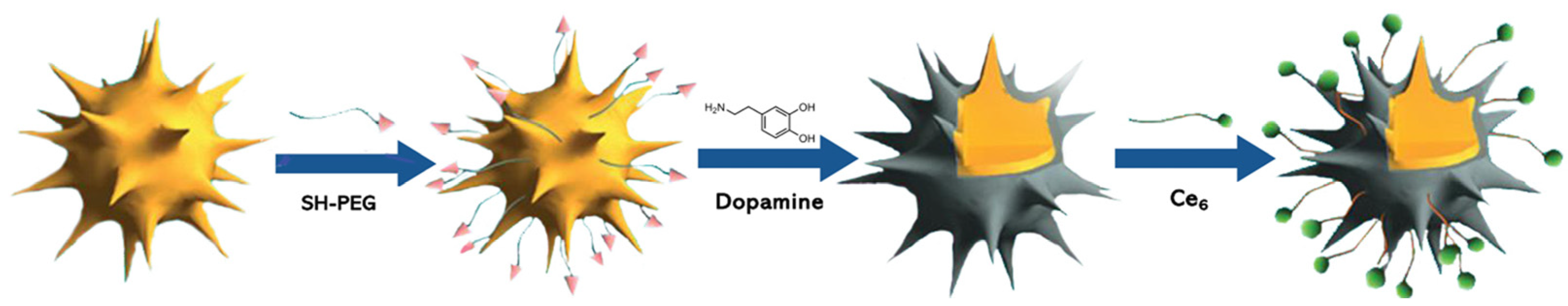{kind=link}
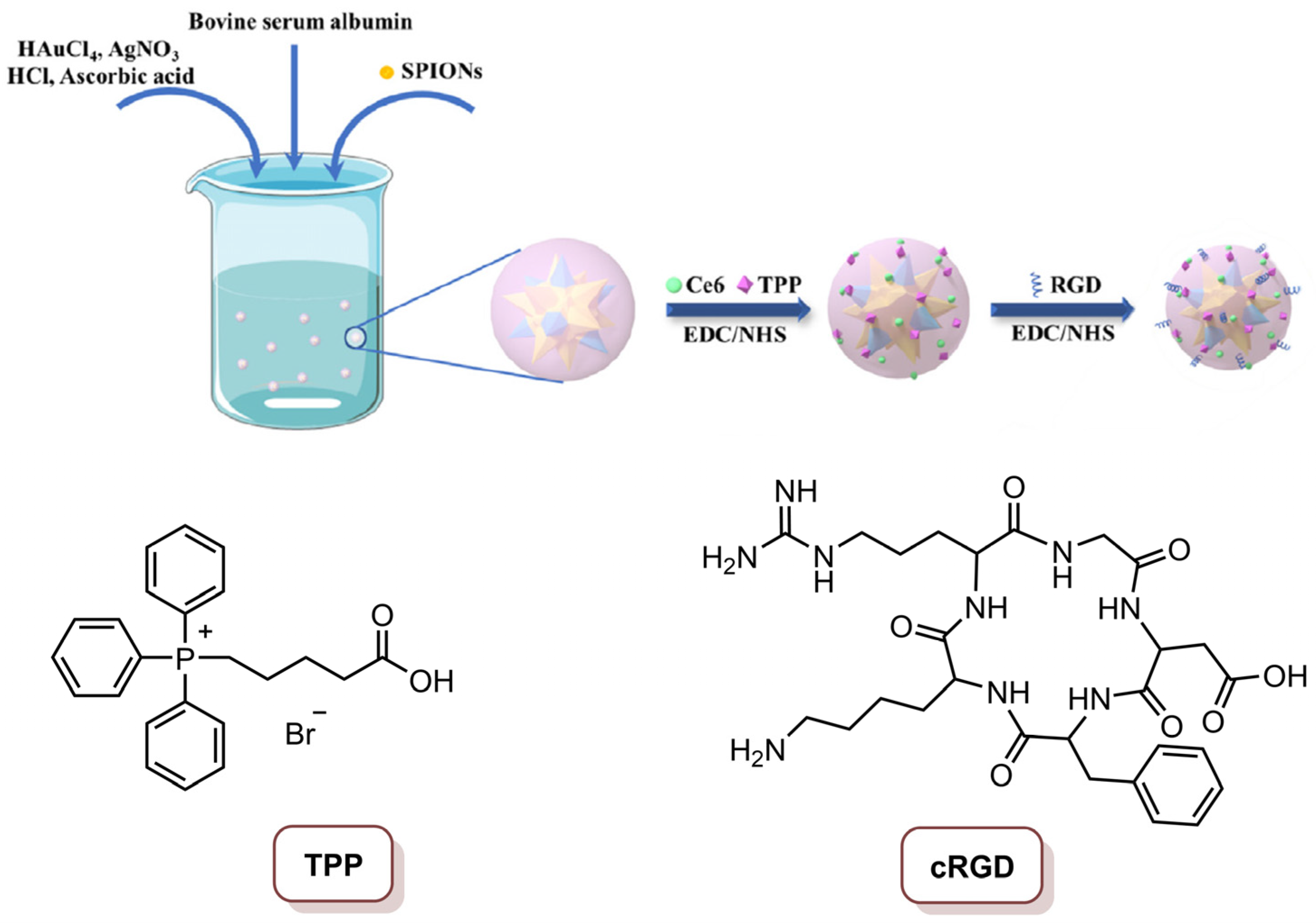{kind=link}
| Photoactive Compund/ Formulation | Chlorin Molecule | Cell Lines | Therapeutic Modality | Light Conditions | Main Findings (IC50 or % Cell/Tumour Inhibition) | Ref. |
|---|---|---|---|---|---|---|
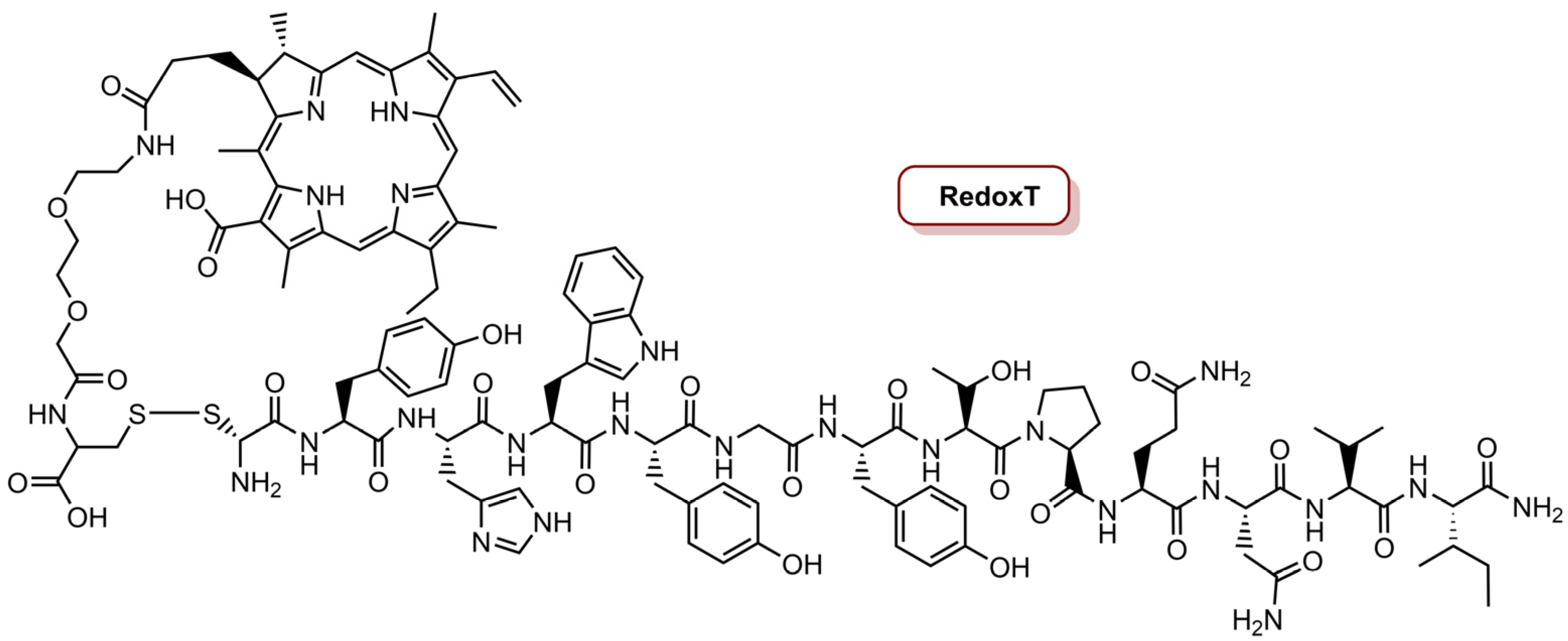
| RedoxT | Ce4 | MDA-MB-231 and MDA-MB-468 | PDT | 670 nm CW laser, 50 W cm−2 (20 J cm−2) | IC50 of 0.66 µM in MDA-MB-468 cells IC50 of 1.80 µM in MDA-MB-231 cells | [75] |
| NVP/Ce6@NPs | Ce6 | MDA-MB-231 | 660 nm laser, 1000 mW cm−2 (1.8 kJ cm−2) | 89.3% tumour size reduction (4.0 mg kg−1 Ce6, 10 mg kg−1 NVP-BEZ235) | [23] | |
| 2D glycocluster | 660 nm laser, 1000 W cm−2 (900 J cm−2) | Cell viability decreased in the presence of glycoprobe (10 µM glycoprobe/1.0 µM Ce6/40 µM HSA/30 µg mL−1 2D MnO2) | [84] | |||
| POP-Gel | 4T1 | 660 nm laser, 5.0 mW cm−2 (9.0 J cm−2) | IC50 of 0.359 µg mL−1 | [82] | ||
| CBTN and CBTX | MePheo | BT-549 | 650 nm LumaCare LC-122, 16 mW cm−2 (0.96 J cm−2, 1.92 J cm−2, 4.8 J cm−2) | 60% cell inhibition for CBTN at 100 nM (4.8 J cm−2); 77% decrease in cancer cells proliferation for CBTN; 16 % decrease for CBX at 0.5 µM (0.96 J cm−2) | [83] | |
| EAT@NPs | Ce6 | MDA-MB-231 | PDT/Chemo | 670 nm CW laser, 68 mW cm−2 (10 J cm−2) | 98% cell inhibition (20 μM CPT equivalent) | [74] |
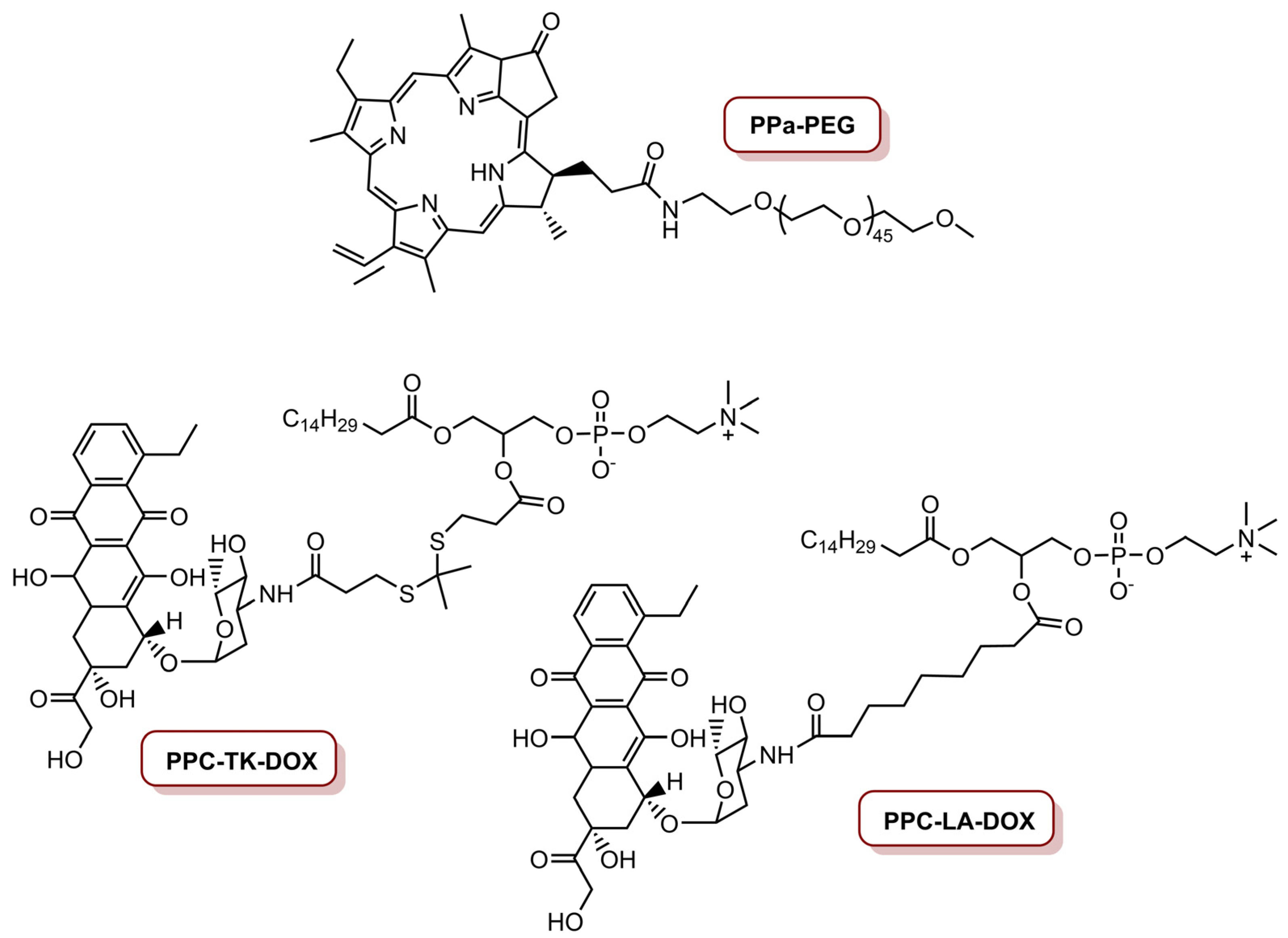
| ELTSL-HOC/DOX | PPa | MDA-MB-231, MDA-MB-468 and 4T1 | 670 nm laser, 400 mW cm−2 (240 J cm−2) | Supressed the growth of the tumour in an orthotopic 4T1 tumour model | [77] | |
| RADV | 4T1 | 670 nm laser, 100 mW cm−2 (12 J cm−2) in vitro; 200 mW cm−2 (60 J cm−2) in vivo | In vitro: 87.5% cell inhibition (DOX concentration of 1.0 µM) In vivo: 85% tumour inhibition (DOX dosage of 5.0 mg kg−1) | [78] | ||
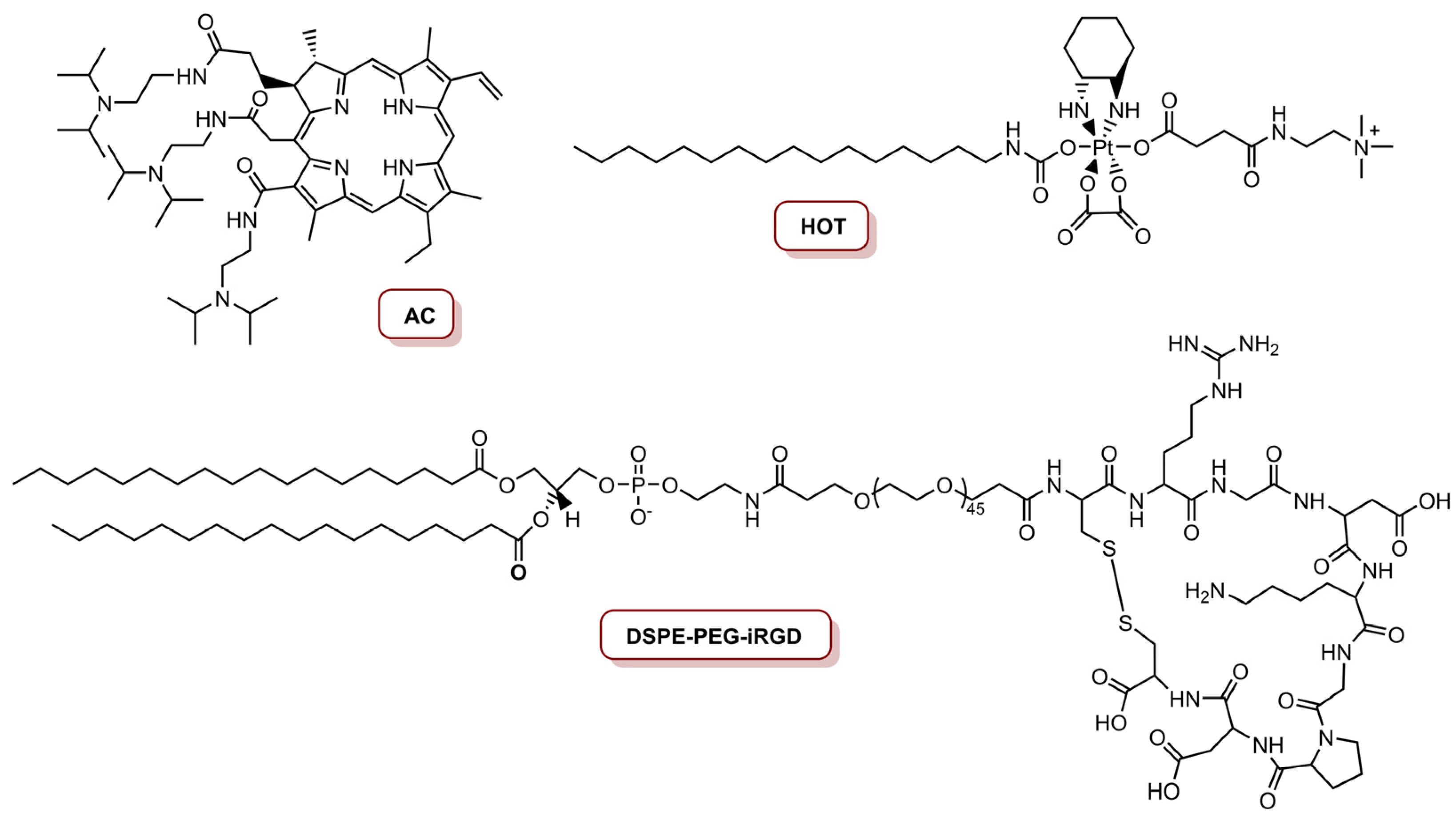
| iNP@AC | Acid derivative of Ce6 (AC) | 655 nm laser, 250 mW cm−2 (7.5 J cm−2), 630 mW cm−2 (18.9 J cm−2) | 81% cell inhibition (concentration of 300 nM platinum and 8.0 nM AC) | [76] | ||
| Ce6@PEICuS NPs | Ce6 | MDA-MB-231 | PDT/PTT | 670 nm laser, 100 mW cm−2 (60 J cm−2); 808 nm laser, 2.0 W cm−2 (1.2 kJ cm−2) | PDT: 55% cell inhibition; PTT: 41% cell inhibition; PDT/PDT: 84% cell inhibition (200 µg mL−1) | [79] |
| Ce6@MGN@RT | 660 nm laser, 30 mW cm−2 (5.4 J cm−2); 808 nm laser, 0.8 W cm−2 (144 J cm−2) | 81% cell inhibition after PDT/PTT (20 µg mL−1) | [22] | |||
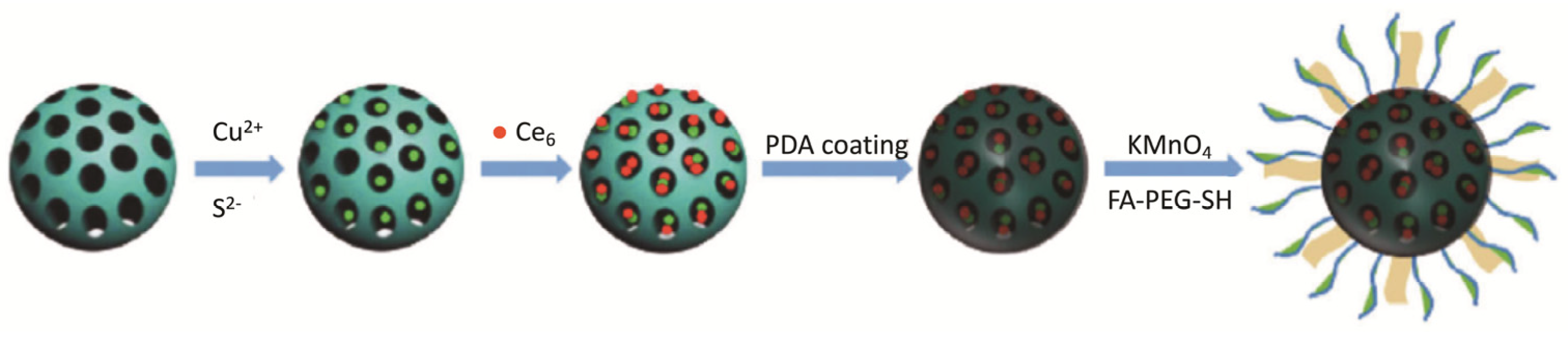
| Ce6-CuS/MSN@PDA@MnO2-FA NPs | 4T1 | 660 nm laser, 50 mW cm−2 (30 J cm−2); 808 nm laser, 2.0 W cm−2 (1.2 kJ cm−2) | 2% of cell viability (16 µg mL−1 of Ce6 and 60 µg mL−1 of CuS) | [85] | ||
| Ce6-PDA@AuNSs | 635 nm laser, 50 mW cm−2 (15 J cm−2); 808 nm laser 1.0 W cm−2 (300 J cm−2) | PDT: reduction to 28.2%; PTT: reduction to 14.7%; PDT/PTT: reduction to approximately zero in cell viability (50 µg mL−1); the tumour disappeared after PDT/PTT treatment (200 µg mL−1) | [86] | |||
| CDTN | Ce6 | 4T1 | PDT/Chemo/siRNA | 671 nm laser, 450 mW cm−2 (27 J cm−2); 110 mW cm−2 (6.6 J cm−2) | Cancer cells were killed in superficial tumours via PDT, and in deep tumours via PDT-potentiated chemotherapy and Twist downregulation | [80] |
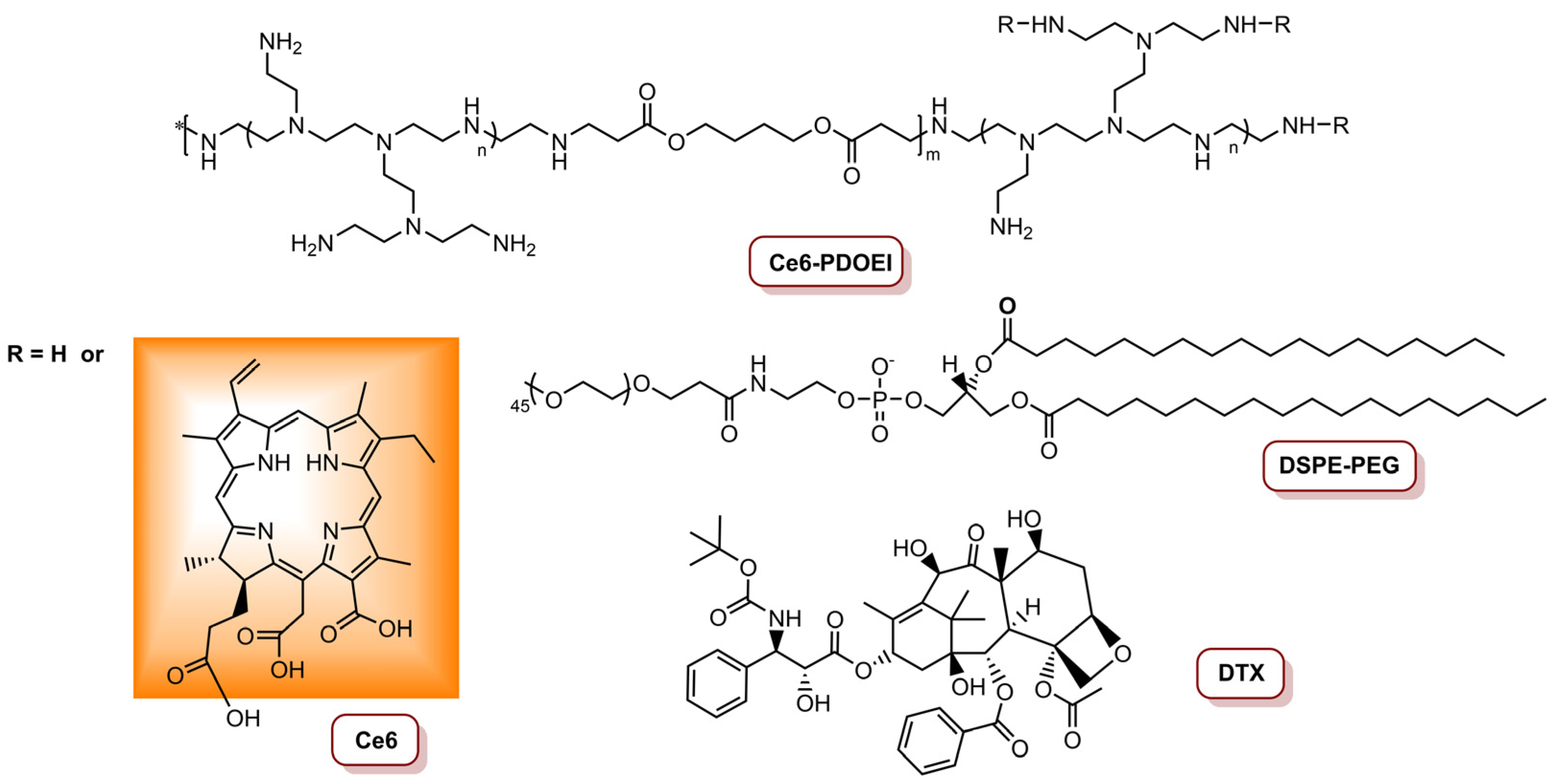
| PCM/DOX/Ce6@H-CuS NPs | Ce6 | 4T1 | PDT/PTT/Chemo | 660 nm laser, 500 mW cm−2 (150 J cm−2); 808 nm laser, 2.0 W cm−2 (600 J cm−2) | 98.4% tumour inhibition (DOX dosage 2.0 mg kg−1 and Ce6 dosage 5.0 mg kg−1) | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dias, C.J.; Helguero, L.; Faustino, M.A.F. Current Photoactive Molecules for Targeted Therapy of Triple-Negative Breast Cancer. Molecules 2021, 26, 7654. https://doi.org/10.3390/molecules26247654
Dias CJ, Helguero L, Faustino MAF. Current Photoactive Molecules for Targeted Therapy of Triple-Negative Breast Cancer. Molecules. 2021; 26(24):7654. https://doi.org/10.3390/molecules26247654
Chicago/Turabian StyleDias, Cristina J., Luisa Helguero, and Maria Amparo F. Faustino. 2021. "Current Photoactive Molecules for Targeted Therapy of Triple-Negative Breast Cancer" Molecules 26, no. 24: 7654. https://doi.org/10.3390/molecules26247654
APA StyleDias, C. J., Helguero, L., & Faustino, M. A. F. (2021). Current Photoactive Molecules for Targeted Therapy of Triple-Negative Breast Cancer. Molecules, 26(24), 7654. https://doi.org/10.3390/molecules26247654