
Recent Advances in the Synthesis of β-Carboline Alkaloids




Abstract
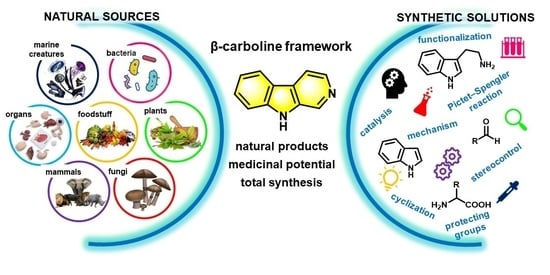
:
1. Introduction
2. Natural Products Containing a Simple β-Carboline Skeleton
3. Fused Ring Systems Bearing a β-Carboline Skeleton
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 9-BNN | 9-borabicyclo[3.3.1]nonane |
| Å | angstrom |
| Ac | acetyl |
| ACCN | 1,1′-azobis(cyclohexanecarbo-nitrile) |
| AIBN | azobisisobutyronitrile |
| Alloc | allyloxycarbonyl |
| aq | aqueous |
| Barton’s base | 2-tert-butyl-1,1,3,3-tetra-methylguanidine |
| Bn | benzyl |
| Boc | tert-butyloxycarbonyl |
| brsm | yield based on recovered starting material |
| BuLi | n-butyllithium |
| Bz | benzoyl |
| CAN | cerium ammonium nitrate |
| cap | caprolactamate |
| CDI | carbonyldiimidazole |
| c-Hex2BH | dicyclohexylborane |
| cod | cyclooctadiene |
| conc. | concentrated |
| Cp2TiMe2 | bis(η5-cyclopentadienyl)-dimethyl-titanium |
| CuDPP | copper(I)-diphenyl phosphinate |
| d | day |
| d.r. | diastereomeric ratio |
| DABCO | 1,4-diazabicyclo[2.2.2]octane |
| dba | dibenzylideneacetone |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DBN | 1,5-diazabicyclo[4.3.0]non-5-ene |
| DCC | N,N′-dicyclohexylcarbodiimide |
| DDQ | 2,3-dichloro-5,6-dicyano-1,4-benzoquinone |
| DEAD | diethyl azodicarboxylate |
| DHβC | 3,4-dihydro-β-carboline |
| DIBAL | diisobutylaluminum hydride |
| DIPEA | N,N-diisopropylethylamine |
| DMAP | 4-dimethylaminopyridine |
| DME | 1,2-dimethoxyethane |
| DMF | dimethylformamide |
| DMP | Dess–Martin periodinane |
| DMS | dimethyl sulfide |
| DMSO | dimethyl sulfoxide |
| e.e. | enantiomeric excess |
| e.r. | enantiomeric ratio |
| EDCl | 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide |
| Et | ethyl |
| h | hour |
| HFIP | 1,1,1,3,3,3-hexafluoroisopropyl alcohol |
| HOBt | 1-hydroxybenzotriazole |
| IBA | 2-iodosobenzoic acid |
| IBX | 2-iodoxybenzoic acid |
| KHMDS | potassium bis(trimethylsilyl)-amide |
| Lawesson’s reagent | 2,4-bis(4-methoxy-phenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide |
| LDA | lithium diisopropylamide |
| LiHMDS | lithium bis(trimethylsilyl)amide |
| L-selectride | lithium tri-sec-butyl(hydrido)-borate |
| m-CPBA | meta-chloroperbenzoic acid |
| Me | methyl |
| MEM | 2-methoxyethoxymethyl ether |
| min. | minute |
| MS | molecular sieve |
| MW | microwave |
| Nap | 2-naphthylmethyl ether |
| NB | 2-nitrobenzyl |
| NBS | N-bromosuccinimide |
| NCS | N-chlorosuccinimide |
| NMM | N-methylmorpholine |
| NMO | 4-methylmorpholine N-oxide |
| NMP | N-methyl-2-pyrrolidone |
| Ns | 2-nitrobenzenesulfonyl |
| o-NBOH | o-nitrobenzylalcohol |
| Oxone® | 2 KHSO5·KHSO4·K2SO4 |
| PDC | pyridinium dichromate |
| Pf | phenylfluorenyl |
| Phth | phthaloyl |
| pin | pinacolato |
| PMB | p-methoxybenzyl |
| Pr | propyl |
| psi | pound-force per square inch |
| Py | pyridine |
| r.t. | room temperature |
| Schwartz’s reagent | chloridobis(η5-cyclo-pentadienyl)hydridozirconium |
| T3P® | propylphosphonic anhydride |
| TBAF | tetrabutylammonium fluoride |
| TBAT | tetrabutylammonium difluoro-triphenylsilicate |
| TBDPS | tert-butyldiphenylsilyl |
| TBDMS | tert-butyldimethylsilyl |
| t-Bu | tert-butyl |
| TES | triethylsilane |
| TMG | tetramethylguanidine |
| Tf | trifluoromethanesulfonyl |
| TFA | trifluoroacetic acid |
| TFAA | trifluoroacetic anhydride |
| THF | tetrahydrofuran |
| THβC | 1,2,3,4-tetrahydro-β-carboline |
| TIPS | triisopropylsilyl |
| TMG | 1,1,3,3-tetramethylguanidine |
| TMP | 2,2,6,6-tetramethylpiperidine |
| TMS | trimethylsilyl |
| TPAP | tetrapropylammonium perruthenate |
| Ts | p-toluenesulfonyl (tosyl) |
| Tsdpen | N-tosyl-1,2-diphenylethylene-1,2-diamine |
References
- Smirnova, O.B.; Golovko, T.V.; Granik, V.G. Carbolines. Part 2: Comparison of some of the properties of α-, γ-, and δ-carbolines. Pharm. Chem. J. 2011, 45, 389–400. [Google Scholar] [CrossRef]
- Singh, V.; Batra, S. 1-Formyl-9H-β-carboline: A useful scaffold for synthesizing substituted and fused β-carbolines. Curr. Org. Synth. 2012, 9, 513–528. [Google Scholar] [CrossRef]
- Piechowska, P.; Zawirska-Wojtasiak, R.; Mildner-Szkudlarz, S. Bioactive β-carbolines in food: A review. Nutrients 2019, 11, 814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Dan, W.; Schneider, U.; Wang, J. β-Carboline alkaloid monomers and dimers: Occurrence, structural diversity, and biological activities. Eur. J. Med. Chem. 2018, 157, 622–656. [Google Scholar] [CrossRef] [PubMed]
- Casal, S. Chapter 20. Potential effects of β-carbolines on human health. In Coffee: Consumption and Health Implications; The Royal Society of Chemistry: Croydon, UK, 2019; pp. 461–468. [Google Scholar]
- Cao, R.; Peng, W.; Wang, Z.; Xu, A. b-Carboline alkaloids: Biochemical and pharmacological functions. Curr. Med. Chem. 2007, 14, 479–500. [Google Scholar] [CrossRef]
- Maity, P.; Adhikari, D.; Jana, A.K. An overview on synthetic entries to tetrahydro-β-carbolines. Tetrahedron 2019, 75, 965–1028. [Google Scholar] [CrossRef]
- Domínguez, G.; Pérez-Castells, J. Chemistry of β-carbolines as synthetic intermediates. Eur. J. Org. Chem. 2011, 2011, 7243–7253. [Google Scholar] [CrossRef]
- Devi, N.; Kumar, S.; Pandey, S.K.; Singh, V. 1(3)-Formyl-β-carbolines: Potential aldo-X precursors for the synthesis of β-carboline-based molecular architectures. Asian J. Org. Chem. 2018, 7, 6–36. [Google Scholar] [CrossRef]
- Inoue, S.; Okada, K.; Tanino, H.; Kakoi, H.; Goto, T. Trace characterization of the fluorescent substances of a dinoflagellate, Noctiluca miliaris. Chem. Lett. 1980, 9, 297–298. [Google Scholar] [CrossRef] [Green Version]
- Herraiz, T. Identification and occurrence of β-carboline alkaloids in raisins and inhibition of monoamine oxidase (MAO). J. Agric. Food. Chem. 2007, 55, 8534–8540. [Google Scholar] [CrossRef]
- Zhu, Y.-Y.; Li, X.; Yu, H.-Y.; Xiong, Y.-F.; Zhang, P.; Pi, H.-F.; Ruan, H.-L. Alkaloids from the bulbs of Lycoris longituba and their neuroprotective and acetylcholinesterase inhibitory activities. Arch. Pharmacal Res. 2015, 38, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.-P.; Zou, Z.-X.; Zhang, Y.; Li, J.; Cheng, F.; Xu, P.-S.; Zhou, G.; Li, X.-M.; Xu, K.-P.; Tan, G.-S. New adenine analogues and a pyrrole alkaloid from Selaginella delicatula. Nat. Prod. Res. 2019, 33, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, N.; Bai, Z.; Di, J.; Zhang, H.; Dong, P.; Zhang, P. Chemical constituent from the peel of Trichosanthes kirilowii Maxim and their NF-κB inhibitory activity. Nat. Prod. Res. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nasser, A.M.; Court, W.E. Leaf alkaloids of Rauwolfia caffra. Phytochemistry 1983, 22, 2297–2300. [Google Scholar] [CrossRef]
- Schumacher, R.W.; Davidson, B.S. Didemnolines A-D, new N9-substituted β-carbolines from the marine ascidian Didemnum sp. Tetrahedron 1995, 51, 10125–10130. [Google Scholar] [CrossRef]
- Rajemiarimoelisoa, C.F.; Boyère, C.; Pellissier, L.; Peuchmaur, M.; Randrianarivo, H.R.; Rakoto, D.A.D.; Jeannoda, V.L.; Boumendjel, A. Chemical composition of the pods of Albizia polyphylla. Nat. Prod. Res. 2016, 30, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Tulyaganov, T.; Kozimova, N.; Allaberdiev, F.K. Alkaloids from plants of the genus Nitraria. Chem. Nat. Compd. 2006, 42, 198–200. [Google Scholar] [CrossRef]
- Badger, G.M.; Beecham, A.F. Isolation of tetrahydroharman from Petalostyles labicheoides. Nature 1951, 168, 517–518. [Google Scholar] [CrossRef]
- Tran, T.D.; Pham, N.B.; Ekins, M.; Hooper, J.N.A.; Quinn, R.J. Isolation and total synthesis of stolonines A–C, unique taurine amides from the australian marine tunicate Cnemidocarpa stolonifera. Mar. Drugs 2015, 13, 4556–4575. [Google Scholar] [CrossRef] [Green Version]
- Hudson, J.B.; Saboune, H.; Abramowski, Z.; Towers, G.H.; Rinehart, K.L., Jr. The photoactive antimicrobial properties of eudistomins from the caribbean tunicate Eudistoma olivaceum. Photochem. Photobiol. 1988, 47, 377–381. [Google Scholar] [CrossRef]
- Tarzi, O.I.; Erra-Balsells, R. Photochemistry of the alkaloids eudistomin N (6-bromo-nor-harmane) and eudistomin O (8-bromo-nor-harmane) and other bromo-β-carbolines. J. Photochem. Photobiol. B Biol. 2005, 80, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Badre, A.; Boulanger, A.; Abou-Mansour, E.; Banaigs, B.; Combaut, G.; Francisco, C. Eudistomin U and isoeudistomin U, new alkaloids from the carribean ascidian Lissoclinum fragile. J. Nat. Prod. 1994, 57, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Kinzer, K.F.; Cardellina, J.H. Three new β-carbolines from the bermudian tunicate Eudistoma olivaceum. Tetrahedron Lett. 1987, 28, 925–926. [Google Scholar] [CrossRef]
- Rajesh, R.P.; Murugan, A. Spectroscopic identification of brominated, non-brominated alkaloids and evaluation of antimicrobial activity of Eudistomin-I, Eudistomin H, from green ascidian Eudistoma viride. J. Appl. Pharm. Sci. 2019, 9, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Rinehart, K.L., Jr. Eudistomins A, D, G, H, I, J, M, N, O, P, and Q, bromo, hydroxy, pyrrolyl and iminoazepino. beta-Carbolines from the antiviral caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1984, 106, 1526–1528. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Kobayashi, J.; Harbour, G.C.; Gilmore, J.; Mascal, M.; Holt, T.G.; Shield, L.S.; Lafargue, F. Eudistomins A-Q,. beta-Carbolines from the antiviral caribbean tunicate Eudistoma olivaceum. J. Am. Chem. Soc. 1987, 109, 3378–3387. [Google Scholar] [CrossRef]
- Kim, D.-C.; Quang, T.H.; Yoon, C.-S.; Ngan, N.T.T.; Lim, S.-I.; Lee, S.-Y.; Kim, Y.-C.; Oh, H. Anti-neuroinflammatory activities of indole alkaloids from kanjang (Korean fermented soy source) in lipopolysaccharide-induced BV2 microglial cells. Food Chem. 2016, 213, 69–75. [Google Scholar] [CrossRef]
- Picker, K.; Ritchie, E.; Taylor, W. The chemical constituents of australian Flindersia species. XXI. An examination of the bark and the leaves of F. laevicarpa. Aust. J. Chem. 1976, 29, 2023–2036. [Google Scholar] [CrossRef]
- Guo, E.; Hu, Y.; Du, T.; Zhu, H.; Chen, L.; Qu, W.; Zhang, J.; Xie, N.; Liu, W.; Feng, F.; et al. Effects of Picrasma quassioides and its active constituents on Alzheimer’s disease in vitro and in vivo. Bioorg. Chem. 2019, 92, 103258. [Google Scholar] [CrossRef]
- Shi, C.-C.; Chen, S.-Y.; Wang, G.-J.; Liao, J.-F.; Chen, C.-F. Vasorelaxant effect of harman. Eur. J. Pharmacol. 2000, 390, 319–325. [Google Scholar] [CrossRef]
- Miralles, A.; Esteban, S.; Sastre-Coll, A.; Moranta, D.; Asensio, V.J.; García-Sevilla, J.A. High-affinity binding of β-carbolines to imidazoline I2B receptors and MAO-A in rat tissues: Norharman blocks the effect of morphine withdrawal on DOPA/noradrenaline synthesis in the brain. Eur. J. Pharmacol. 2005, 518, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Moty, S.; Sakai, S.; Aimi, N.; Takayama, H.; Kitajima, M.; El-Shorbagi, A.; Ahmed, A.; Omar, N. Synthesis of cytotoxic 1-polyhydroxyalkyl-β-carboline derivatives. Eur. J. Med. Chem. 1998, 32, 1009–1017. [Google Scholar] [CrossRef]
- Sung, Y.; Koike, K.; Nikaido, T.; Ohmoto, T.; Sankawa, U. Inhibitors of cyclic AMP phosphodiesterase in Picrasma quassioides Bennet, and inhibitory activities of related beta-carboline alkaloids. Chem. Pharm. Bull. 1984, 32, 1872–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, W.H.; Gao, H.; Zhao, F.; Lin, H.W.; Pan, Y.M.; Zhou, G.X.; Yao, X.S. Anti-inflammatory alkaloids from the stems of Picrasma quassioides Bennet. Chem. Pharm. Bull. 2011, 59, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Li, H.; Luan, R.; Huang, G.; Liu, Y.; Wang, M.; Chao, Q.; Wang, L.; Li, D.; Fan, H.; et al. Identification of β-carboline and canthinone alkaloids as anti-inflammatory agents but with different inhibitory profile on the expression of iNOS and COX-2 in lipopolysaccharide-activated RAW 264.7 macrophages. J. Nat. Med. 2019, 73, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Li, S.-F.; Zhang, Y.; Li, Y.; Li, X.-R.; Kong, L.-M.; Tan, C.-J.; Li, S.-L.; Di, Y.-T.; He, H.-P.; Hao, X.-J. β-Carboline alkaloids from the leaves of Trigonostemon lii. Bioorg. Med. Chem. Lett. 2012, 22, 2296–2299. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, P.; Kong, F.D.; Wang, J.; Zuo, W.J.; Wang, H.; Dai, H.F.; Mei, W.L. Two new alkaloids from the twigs of Trigonostemon filipes. J. Asian Nat. Prod. Res. 2018, 20, 270–276. [Google Scholar] [CrossRef]
- Machowinski, A.; Krämer, H.J.; Hort, W.; Mayser, P. Pityriacitrin—A potent UV filter produced by Malassezia furfur and its effect on human skin microflora. Mycoses 2006, 49, 388–392. [Google Scholar] [CrossRef]
- Nagao, T.; Adachi, K.; Nishida, F.; Nishishima, M.; Mochida, K. New Ultraviolet-Absorbing Substance Produced by Marine Bacteria and Its Production. JP Patent 11,269,175, 5 October 1999. [Google Scholar]
- Chen, Y.-X.; Xu, M.-Y.; Li, H.-J.; Zeng, K.-J.; Ma, W.-Z.; Tian, G.-B.; Xu, J.; Yang, D.-P.; Lan, W.-J. Diverse secondary metabolites from the marine-derived fungus Dichotomomyces cejpii F31-1. Mar. Drugs 2017, 15, 339. [Google Scholar] [CrossRef] [Green Version]
- Salmoun, M.; Devijver, C.; Daloze, D.; Braekman, J.-C.; van Soest, R.W. 5-Hydroxytryptamine-derived alkaloids from two marine sponges of the genus Hyrtios. J. Nat. Prod. 2002, 65, 1173–1176. [Google Scholar] [CrossRef]
- Huang, W.; Yi, X.; Feng, J.; Wang, Y.; He, X. Piperidine alkaloids from Alocasia macrorrhiza. Phytochemistry 2017, 143, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Li, S.F.; Cheng, Y.Y.; Zhang, Y.; Li, S.L.; He, H.P.; Hao, X.J. β-carboline alkaloids from Trigonostemon filipes and Trigonostemon lii. Nat. Prod. Bioprospect. 2012, 2, 126–129. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Sun, X.; Xu, B.; Bijian, K.; Wan, S.; Li, G.; Alaoui-Jamali, M.; Jiang, T. Total synthesis and bioactivity of the marine alkaloid pityriacitrin and some of its derivatives. Eur. J. Med. Chem. 2011, 46, 6089–6097. [Google Scholar] [CrossRef] [PubMed]
- Liew, L.P.; Fleming, J.M.; Longeon, A.; Mouray, E.; Florent, I.; Bourguet-Kondracki, M.-L.; Copp, B.R. Synthesis of 1-indolyl substituted β-carboline natural products and discovery of antimalarial and cytotoxic activities. Tetrahedron 2014, 70, 4910–4920. [Google Scholar] [CrossRef]
- Mexia, N.; Gaitanis, G.; Velegraki, A.; Soshilov, A.; Denison, M.S.; Magiatis, P. Pityriazepin and other potent AhR ligands isolated from Malassezia furfur yeast. Arch. Biochem. Biophys. 2015, 571, 16–20. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.-h.; Chen, C.; Wang, H.; Ye, W.-c.; Zhou, G.-x. Indole alkaloids from Alocasia macrorrhiza. Chem. Pharm. Bull. 2012, 60, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.D.; da Silva, T.; Aguiar, A.C.C.; Oliva, G.; Guido, R.V.; Yokoyama-Yasunaka, J.K.; Uliana, S.R.; Lopes, L.M. Chemical composition, antiprotozoal and cytotoxic activities of indole alkaloids and benzofuran neolignan of Aristolochia cordigera. Planta Med. 2017, 83, 912–920. [Google Scholar] [CrossRef]
- Sauleau, P.; Martin, M.-T.; Dau, M.-E.T.H.; Youssef, D.T.; Bourguet-Kondracki, M.-L. Hyrtiazepine, an azepino-indole-type alkaloid from the Red Sea marine sponge Hyrtios erectus. J. Nat. Prod. 2006, 69, 1676–1679. [Google Scholar] [CrossRef]
- Diallo, A.O.; Mehri, H.; Iouzalen, L.; Plat, M. Alkaloids from leaves of Alangium bussyanum. Phytochemistry 1995, 40, 975–977. [Google Scholar] [CrossRef]
- Patil, A.D.; Freyer, A.J.; Carte, B.; Taylor, P.B.; Johnson, R.K.; Faulkner, D.J. Haploscleridamine, a novel tryptamine-derived alkaloid from a aponge of the order Haplosclerida: An inhibitor of Cathepsin K. J. Nat. Prod. 2002, 65, 628–629. [Google Scholar] [CrossRef]
- Wang, K.B.; Li, S.G.; Huang, X.Y.; Li, D.H.; Li, Z.L.; Hua, H.M. (±)-Peharmaline A: A pair of rare β-carboline–vasicinone hybrid alkaloid enantiomers from Peganum harmala. Eur. J. Org. Chem. 2017, 2017, 1876–1879. [Google Scholar] [CrossRef]
- Wang, S.G.; Xia, Z.L.; Xu, R.Q.; Liu, X.J.; Zheng, C.; You, S.L. Construction of chiral tetrahydro-β-carbolines: Asymmetric Pictet–Spengler reaction of indolyl dihydropyridines. Angew. Chem. 2017, 129, 7548–7551. [Google Scholar] [CrossRef]
- Tulyaganov, T.S.; Nazarov, O.M.; Levkovich, M.G.; Abdullaev, N.D. Alkaloids of the Nitraria genus. Komavine and Acetylkomavine. Chem. Nat. Compd. 2001, 37, 61–64. [Google Scholar] [CrossRef]
- Huang, H.; Yao, Y.; He, Z.; Yang, T.; Ma, J.; Tian, X.; Li, Y.; Huang, C.; Chen, X.; Li, W. Antimalarial β-carboline and indolactam alkaloids from Marinactinospora thermotolerans, a deep sea isolate. J. Nat. Prod. 2011, 74, 2122–2127. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tang, Y.; Jin, H.J.; Cui, Y.D.; Zhang, L.J.; Jiang, T. An efficient synthesis method targeted to marine alkaloids marinacarbolines A-D and their antitumor activities. J. Asian Nat. Prod. Res. 2015, 17, 299–305. [Google Scholar] [CrossRef]
- Jaeger, R.J.; Lamshöft, M.; Gottfried, S.; Spiteller, M.; Spiteller, P. HR-MALDI-MS imaging assisted screening of β-carboline alkaloids discovered from Mycena metata. J. Nat. Prod. 2013, 76, 127–134. [Google Scholar] [CrossRef]
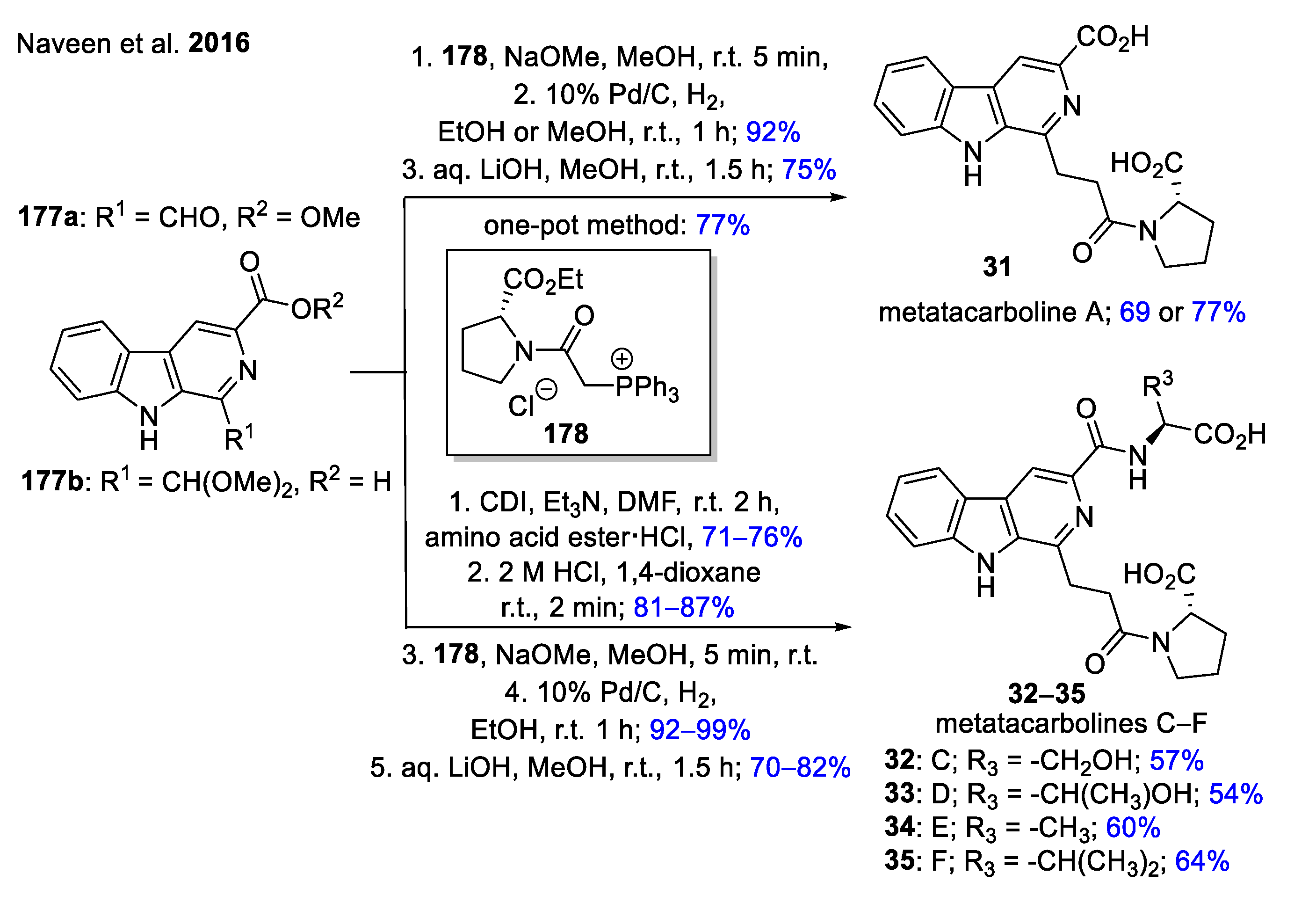
- Naveen, B.; Mudiraj, A.; Khamushavalli, G.; Babu, P.P.; Nagarajan, R. Concise total synthesis of water soluble metatacarboline A, C, D, E and F and its anticancer activity. Eur. J. Med. Chem. 2016, 113, 167–178. [Google Scholar] [CrossRef]
- Oku, N.; Matsunaga, S.; Fusetani, N. Shishijimicins A−C, novel enediyne antitumor antibiotics from the ascidian Didemnum proliferum. J. Am. Chem. Soc. 2003, 125, 2044–2045. [Google Scholar] [CrossRef]
- Ohmoto, T.; Koike, K. Chapter 3 Canthin-6-one Alkaloids. In The Alkaloids: Chemistry and Pharmacology; Brossi, A., Ed.; Academic Press: New York, NY, USA, 1990; Volume 36, pp. 135–170. [Google Scholar]
- Kam, T.-S.; Sim, K.-M. Alkaloids from Kopsia griffithii. Phytochemistry 1998, 47, 145–147. [Google Scholar] [CrossRef]
- Wen-sen, C. The isolation and structure of cordatanine from Drymaria cordata (L.) Willd. J. Integr. Plant Biol. 1986, 28, 450–452. [Google Scholar]
- Hsieh, P.-W.; Chang, F.-R.; Lee, K.-H.; Hwang, T.-L.; Chang, S.-M.; Wu, Y.-C. A new anti-HIV alkaloid, drymaritin, and a new C-glycoside flavonoid, diandraflavone, from Drymaria diandra. J. Nat. Prod. 2004, 67, 1175–1177. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, I.; Allmendinger, L.; Bracher, F. Revised structure of the alkaloid drymaritin. J. Nat. Prod. 2009, 72, 1908–1910. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Genta-Jouve, G.; Zheng, Y.; Zhang, Q.; Chen, C.; Zhou, Q.; Wang, J.; Zhu, H.; Zhang, Y. Griseofamines A and B: Two Indole-Tetramic Acid Alkaloids with 6/5/6/5 and 6/5/7/5 Ring Systems from Penicillium griseofulvum. Org. Lett. 2018, 20, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, Z. Total synthesis and antibacterial activity evaluation of griseofamine A and 16-epi-griseofamine A. Org. Lett. 2019, 21, 2393–2396. [Google Scholar] [CrossRef] [PubMed]
- Besslièvre, R.; Cosson, B.P.; Das, B.C.; Husson, H.P. Structure and total synthesis of deplancheine, a novel indoloquinolizidine alkaloid. Tetrahedron Lett. 1980, 21, 63–66. [Google Scholar] [CrossRef]
- Shi, B.B.; Chen, J.; Bao, M.F.; Zeng, Y.; Cai, X.H. Alkaloids isolated from Tabernaemontana bufalina display xanthine oxidase inhibitory activity. Phytochemistry 2019, 166, 112060. [Google Scholar] [CrossRef]
- Bao, M.-F.; Zeng, C.-X.; Liu, Y.-P.; Zhang, B.-J.; Ni, L.; Luo, X.-D.; Cai, X.-H. Indole alkaloids from Hunteria zeylanica. J. Nat. Prod. 2017, 80, 790–797. [Google Scholar] [CrossRef]
- Yan, W.; Ge, H.M.; Wang, G.; Jiang, N.; Mei, Y.N.; Jiang, R.; Li, S.J.; Chen, C.J.; Jiao, R.H.; Xu, Q.; et al. Pictet-Spengler reaction-based biosynthetic machinery in fungi. Proc. Natl. Acad. Sci. USA 2014, 111, 18138–18143. [Google Scholar] [CrossRef] [Green Version]
- Khokhar, S.; Feng, Y.; Campitelli, M.R.; Ekins, M.G.; Hooper, J.N.A.; Beattie, K.D.; Sadowski, M.C.; Nelson, C.C.; Davis, R.A. Isolation, structure determination and cytotoxicity studies of tryptophan alkaloids from an Australian marine sponge Hyrtios sp. Bioorg. Med. Chem. Lett. 2014, 24, 3329–3332. [Google Scholar] [CrossRef]
- Rath, B.; Hochmair, M.; Plangger, A.; Hamilton, G. Anticancer activity of fascaplysin against lung cancer cell and small cell lung cancer circulating tumor cell lines. Mar. Drugs 2018, 16, 383. [Google Scholar] [CrossRef] [Green Version]
- Roll, D.M.; Ireland, C.M.; Lu, H.S.M.; Clardy, J. Fascaplysin, an unusual antimicrobial pigment from the marine sponge Fascaplysinopsis sp. J. Org. Chem. 1988, 53, 3276–3278. [Google Scholar] [CrossRef]
- Segraves, N.L.; Robinson, S.J.; Garcia, D.; Said, S.A.; Fu, X.; Schmitz, F.J.; Pietraszkiewicz, H.; Valeriote, F.A.; Crews, P. Comparison of fascaplysin and related alkaloids: A study of structures, cytotoxicities, and sources. J. Nat. Prod. 2004, 67, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Asahina, Y.J.; Kashiwaki, K. Chemical constituents of the fruits of Evodia rutaecarpa. Pharm. Soc. Jpn. 1915, 405, 1293. [Google Scholar]
- Sun, Q.; Xie, L.; Song, J.; Li, X. Evodiamine: A review of its pharmacology, toxicity, pharmacokinetics and preparation researches. J. Ethnopharmacol. 2020, 262, 113164. [Google Scholar] [CrossRef] [PubMed]
- Nasser, A.M.A.G.; Court, W.E. Stem bark alkaloids of rauvolfia caffra. J. Ethnopharmacol. 1984, 11, 99–117. [Google Scholar] [CrossRef]
- Majumdar, S.P.; Potier, P.; Poisson, J. The structure of suaveoline, a new alkaloid from Rauwolfia suaveolens S. Moore (apocynaceae). Tetrahedron Lett. 1972, 13, 1563–1566. [Google Scholar] [CrossRef]
- Amer, M.M.A.; Court, W.E. Stem bark alkaloids of Rauwolfia macrophylla. Planta Med. 1980, 40, 8–12. [Google Scholar] [CrossRef]
- Zhan, G.; Miao, R.; Zhang, F.; Hao, X.; Zheng, X.; Zhang, H.; Zhang, X.; Guo, Z. Monoterpene indole alkaloids with diverse skeletons from the stems of Rauvolfia vomitoria and their acetylcholinesterase inhibitory activities. Phytochemistry 2020, 177, 112450. [Google Scholar] [CrossRef]
- Xu, Y.-J.; Tang, C.-P.; Ke, C.-Q.; Ye, Y. Indole alkaloids from leaves and stems of Hunteria zeylanica. Chem. Nat. Compd. 2009, 45, 834–836. [Google Scholar] [CrossRef]
- Schnoes, H.K.; Biemann, K.; Mokry, J.; Kompis, I.; Chatterjee, A.; Ganguli, G. Strictamine. J. Org. Chem. 1966, 31, 1641–1642. [Google Scholar] [CrossRef]
- Ahmad, Y.; Fatima, K.; Atta ur, R.; Occolowitz, J.L.; Solheim, B.A.; Clardy, J.; Garnick, R.L.; Le Quesne, P.W. Structure and absolute configuration of strictamine and strictalamine from Rhazya stricta. Stereochemistry of the Picralima alkaloids. J. Am. Chem. Soc. 1977, 99, 1943–1946. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.-J.; Zhang, D.-B.; Wen, J.; Zhao, X.-W.; Li, Y.; Gao, K. An unusual indole alkaloid with anti-adenovirus and anti-HSV activities from Alstonia scholaris. Tetrahedron Lett. 2014, 55, 1815–1817. [Google Scholar] [CrossRef]
- Wong, S.-P.; Chong, K.-W.; Lim, K.-H.; Lim, S.-H.; Low, Y.-Y.; Kam, T.-S. Arborisidine and arbornamine, two monoterpenoid indole alkaloids with new polycyclic carbon–nitrogen skeletons derived from a common pericine precursor. Org. Lett. 2016, 18, 1618–1621. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, T.A.; Verpoorte, R.; Baerheim Svendsen, A. Alkaloids of Tabernaemontana eglandulosa. Tetrahedron 1984, 40, 737–748. [Google Scholar] [CrossRef]
- Gilani, S.A.; Kikuchi, A.; Shinwari, Z.K.; Khattak, Z.I.; Watanabe, K.N. Phytochemical, pharmacological and ethnobotanical studies of Rhazya stricta Decne. Phytother. Res. Int. J. Devoted Pharmacol. Toxicol. Eval. Nat. Prod. Deriv. 2007, 21, 301–307. [Google Scholar] [CrossRef]
- Kitajima, M.; Nakano, S.; Kogure, N.; Subhadhirasakul, S.; Takayama, H. New indole alkaloids from Ervatamia cumingiana (Dedicated to Professor Tohru Fukuyama on the occasion of his 70th birthday). Heterocycles Int. J. Rev. Commun. Heterocycl. Chem. 2019, 99, 213–221. [Google Scholar]
- Ndongo, J.T.; Mbing, J.N.; Monteillier, A.; Tala, M.F.; Rütten, M.; Mombers, D.; Cuendet, M.; Pegnyemb, D.E.; Dittrich, B.; Laatsch, H. Carbazole-, aspidofractinine-, and aspidocarpamine-type alkaloids from Pleiocarpa pycnantha. J. Nat. Prod. 2018, 81, 1193–1202. [Google Scholar] [CrossRef]
- Zhang, B.-J.; Teng, X.-F.; Bao, M.-F.; Zhong, X.-H.; Ni, L.; Cai, X.-H. Cytotoxic indole alkaloids from Tabernaemontana officinalis. Phytochemistry 2015, 120, 46–52. [Google Scholar] [CrossRef]
- Subramaniam, G.; Hiraku, O.; Hayashi, M.; Koyano, T.; Komiyama, K.; Kam, T.-S. Biologically active aspidofractinine alkaloids from Kopsia singapurensis. J. Nat. Prod. 2008, 71, 53–57. [Google Scholar] [CrossRef]
- Lim, S.-H.; Sim, K.-M.; Abdullah, Z.; Hiraku, O.; Hayashi, M.; Komiyama, K.; Kam, T.-S. Leuconoxine, kopsinitarine, kopsijasmine, and kopsinone derivatives from Kopsia. J. Nat. Prod. 2007, 70, 1380–1383. [Google Scholar] [CrossRef]
- Liu, L.; Chen, Y.-Y.; Qin, X.-J.; Wang, B.; Jin, Q.; Liu, Y.-P.; Luo, X.-D. Antibacterial monoterpenoid indole alkaloids from Alstonia scholaris cultivated in temperate zone. Fitoterapia 2015, 105, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Penelle, J.; Tits, M.; Christen, P.; Molgo, J.; Brandt, V.; Frédérich, M.; Angenot, L. Quaternary indole alkaloids from the stem bark of Strychnos guianensis. Phytochemistry 2000, 53, 1057–1066. [Google Scholar] [CrossRef]
- Naaz, H.; Singh, S.; Pandey, V.P.; Singh, P.; Dwivedi, U.N. Anti-cholinergic alkaloids as potential therapeutic agents for Alzheimer’s disease: An in silico approach. Indian J. Biochem. Biophys. 2013, 50, 120–125. [Google Scholar] [PubMed]
- Kitajima, M.; Anbe, M.; Kogure, N.; Wongseripipatana, S.; Takayama, H. Indole alkaloids from Kopsia jasminiflora. Tetrahedron 2014, 70, 9099–9106. [Google Scholar] [CrossRef]
- Kam, T.-S.; Arasu, L.; Yoganathan, K. Alkaloids from Kopsia pauciflora. Phytochemistry 1996, 43, 1385–1387. [Google Scholar] [CrossRef]
- Walser, A.; Djerassi, C. Alkaloid-Studien LII. Die Alkaloide aus Vallesia dichotoma. Helv. Chim. Acta 1965, 48, 391–404. [Google Scholar] [CrossRef]
- Renner, U. Alkaloide aus Schizozygia Caffaeoides. 3. Strukturelle Beziehungen zwischen Schizozygin und einigen Nebenalkaloiden. Lloydia 1964, 27, 406–415. [Google Scholar]
- Wang, K.-B.; Di, Y.-T.; Bao, Y.; Yuan, C.-M.; Chen, G.; Li, D.-H.; Bai, J.; He, H.-P.; Hao, X.-J.; Pei, Y.-H. Peganumine A, a β-carboline dimer with a new octacyclic scaffold from Peganum harmala. Org. Lett. 2014, 16, 4028–4031. [Google Scholar] [CrossRef]
- Neuss, N.; Boaz, H.E.; Forbes, J. Rauwolfia Serpentina alkaloids. I. Structure of reserpine. J. Am. Chem. Soc. 1954, 76, 2463–2467. [Google Scholar] [CrossRef]
- Tsioufis, C.; Thomopoulos, C. Combination drug treatment in hypertension. Pharmacol. Res. 2017, 125, 266–271. [Google Scholar] [CrossRef]
- De Silva, K.T.D.; King, D.; Smith, G.N. 5α-Carboxystrictosidine. J. Chem. Soc. D Chem. Commun. 1971, 16, 908–909. [Google Scholar] [CrossRef]
- Ferrari, F.; Messana, I.; Botta, B.; de Mello, J.F. Constituents of Guettarda platypoda. J. Nat. Prod. 1986, 49, 1150–1151. [Google Scholar] [CrossRef]
- Kitajima, M.; Hashimoto, K.-i.; Yokoya, M.; Takayama, H.; Aimi, N.; Sakai, S.-i. A New gluco indole alkaloid, 3,4-dehydro-5-carboxystrictosidine, from Preruvian Una de Gato (Uncaria tomentosa). Chem. Pharm. Bull. 2000, 48, 1410–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onozawa, T.; Kitajima, M.; Kogure, N.; Peerakam, N.; Santiarworn, D.; Takayama, H. A Cyclopeptide and a tetrahydroisoquinoline ílkaloid from Ophiorrhiza nutans. J. Nat. Prod. 2017, 80, 2156–2160. [Google Scholar] [CrossRef]
- Brown, R.T.; Charalambides, A.A. Adina alkaloids: The structure of rubenine. J. Chem. Soc. Chem. Commun. 1973, 20, 765–766. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Xiang, J.-C.; Cheng, Y.; Ma, J.-T.; Wu, Y.-D.; Wu, A.-X. Direct biomimetic synthesis of β-carboline alkaloids from two amino acids. J. Org. Chem. 2018, 83, 12247–12254. [Google Scholar] [CrossRef]
- Lood, C.; Nieger, M.; Koskinen, A. Enantiospecific gram scale synthesis of (S)-eleagnine. Tetrahedron 2015, 71, 5019–5024. [Google Scholar] [CrossRef]
- Ellman, J.A.; Owens, T.D.; Tang, T.P. N-tert-butanesulfinyl imines: Versatile intermediates for the asymmetric synthesis of amines. Acc. Chem. Res. 2002, 35, 984–995. [Google Scholar] [CrossRef]
- Reddy, N.S.S.; Babu, R.A.; Reddy, B.V.S. Asymmetric synthesis of tetrahydro-β-carboline alkaloids employing Ellman’s chiral auxiliary. Synthesis 2016, 48, 1079–1086. [Google Scholar] [CrossRef]
- Van Maarseveen, J.H.; Hermkens, P.H.; De Clercq, E.; Balzarini, J.; Scheeren, H.W.; Kruse, C.G. Antiviral and antitumor structure-activity relationship studies on tetracyclic eudistomines. J. Med. Chem. 1992, 35, 3223–3230. [Google Scholar] [CrossRef]
- Roggero, C.M.; Giulietti, J.M.; Mulcahy, S.P. Efficient synthesis of eudistomin U and evaluation of its cytotoxicity. Bioorg. Med. Chem. Lett. 2014, 24, 3549–3551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolodina, A.A.; OV, S. Eudistomin U, isoeudistomin U, and related indole compounds: Synthesis and biological activity. Heterocycles 2018, 96, 1171–1196. [Google Scholar] [CrossRef]
- Kamal, A.; Sathish, M.; Prasanthi, A.V.G.; Chetna, J.; Tangella, Y.; Srinivasulu, V.; Shankaraiah, N.; Alarifi, A. An efficient one-pot decarboxylative aromatization of tetrahydro-β-carbolines by using N-chlorosuccinimide: Total synthesis of norharmane, harmane and eudistomins. RSC Adv. 2015, 5, 90121–90126. [Google Scholar] [CrossRef]
- Davis, R.A.; Carroll, A.R.; Quinn, R.J. Eudistomin V, a New β-carboline from the australian ascidian Pseudodistoma aureum. J. Nat. Prod. 1998, 61, 959–960. [Google Scholar] [CrossRef] [PubMed]
- Hati, S.; Sen, S. N-Bromo-succinimide promoted synthesis of β-carbolines and 3,4-dihydro-β-carbolines from tetrahydro-β-carbolines. Tetrahedron Lett. 2016, 57, 1040–1043. [Google Scholar] [CrossRef]
- Kamal, A.; Tangella, Y.; Manasa, K.L.; Sathish, M.; Srinivasulu, V.; Chetna, J.; Alarifi, A. PhI(OAc)2-mediated one-pot oxidative decarboxylation and aromatization of tetrahydro-β-carbolines: Synthesis of norharmane, harmane, eudistomin U and eudistomin I. Org. Biomol. Chem. 2015, 13, 8652–8662. [Google Scholar] [CrossRef] [PubMed]
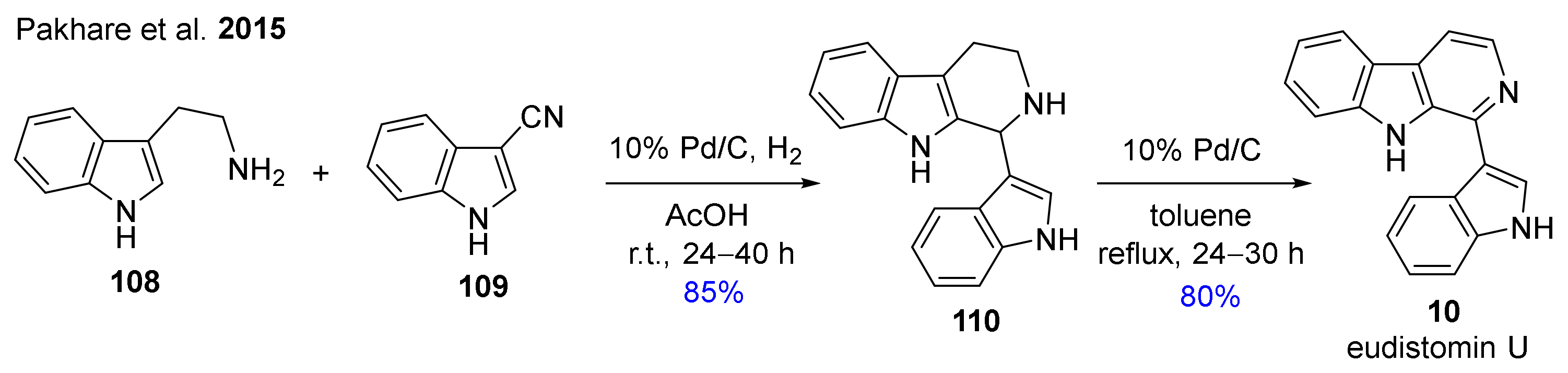
- Pakhare, D.S.; Kusurkar, R.S. Synthesis of tetrahydro-β-carbolines, β-carbolines, and natural products, (±)-harmicine, eudistomin U and canthine by reductive Pictet–Spengler cyclization. Tetrahedron Lett. 2015, 56, 6012–6015. [Google Scholar] [CrossRef]
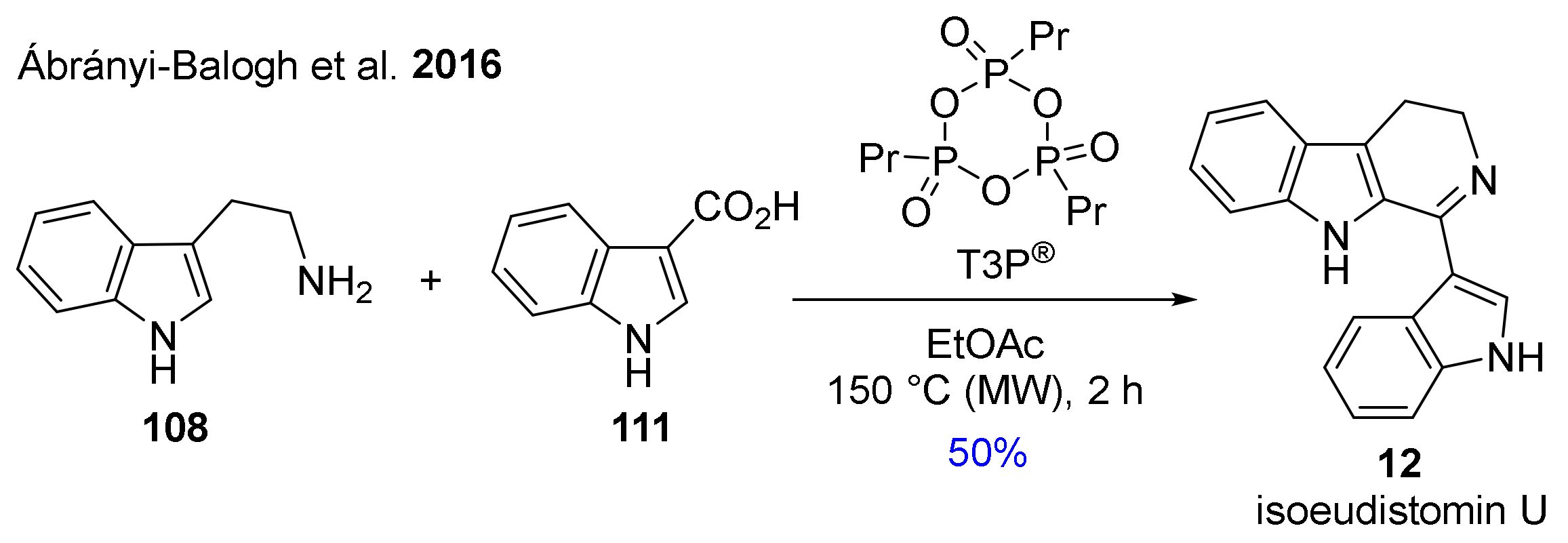
- Ábrányi-Balogh, P.; Földesi, T.; Grün, A.; Volk, B.; Keglevich, G.; Milen, M. Synthetic study on the T3P®-promoted one-pot preparation of 1-substituted-3,4-dihydro-β-carbolines by the reaction of tryptamine with carboxylic acids. Tetrahedron Lett. 2016, 57, 1953–1957. [Google Scholar] [CrossRef]
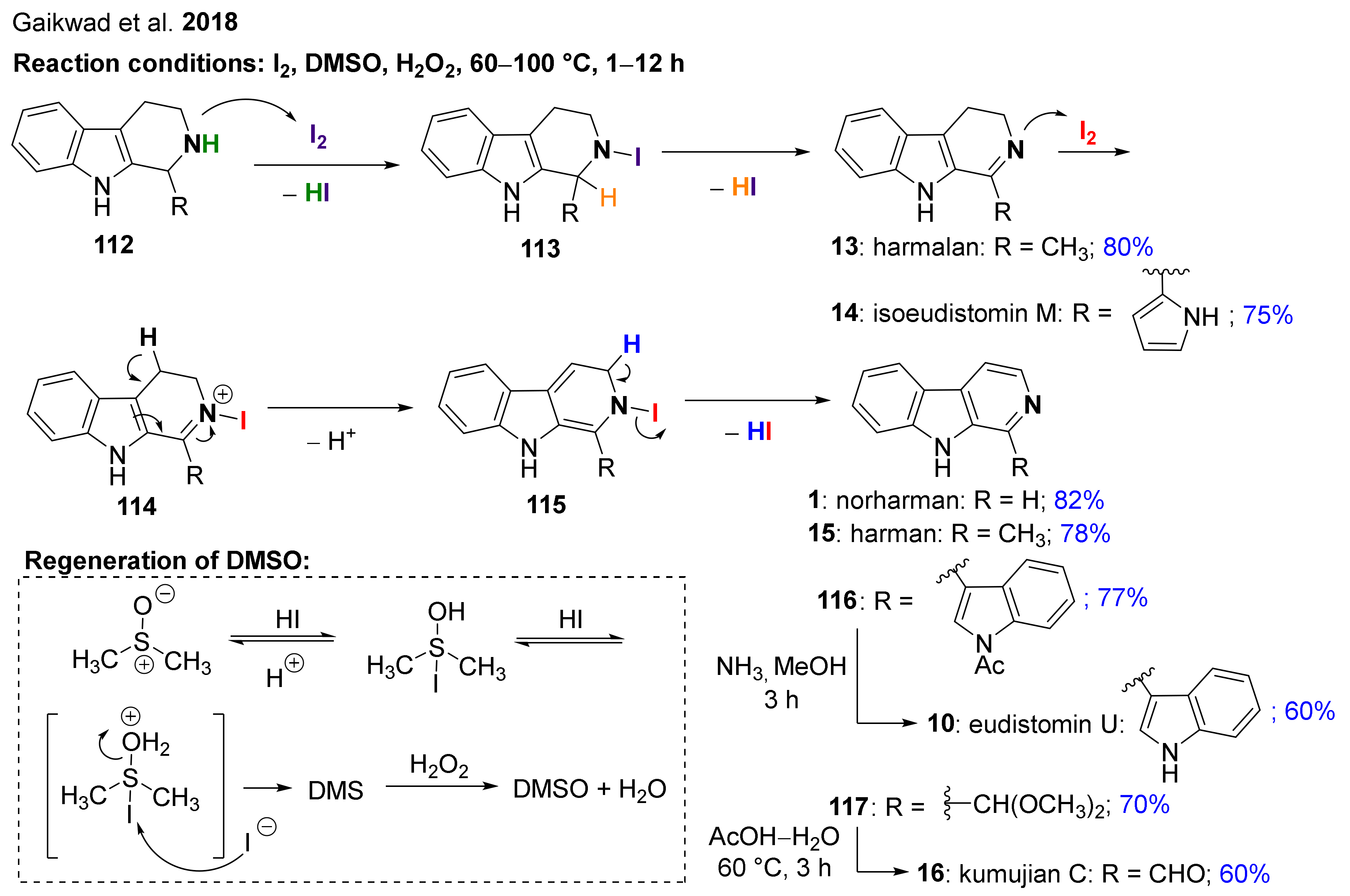
- Gaikwad, S.; Kamble, D.; Lokhande, P. Iodine-catalyzed chemoselective dehydrogenation and aromatization of tetrahydro-β-carbolines: A short synthesis of kumujian-C, eudistomin-U, norharmane, harmane harmalan and isoeudistomine M. Tetrahedron Lett. 2018, 59, 2387–2392. [Google Scholar] [CrossRef]
- Szabó, T.; Dancso, A.; Volk, B.; Milen, M. First total synthesis of beta-carboline alkaloid trigonostemine G and its derivatives. Nat. Prod. Res. 2021, 35, 72–79. [Google Scholar] [CrossRef]
- Szabó, T.; Hazai, V.; Volk, B.; Simig, G.; Milen, M. First total synthesis of the β-carboline alkaloids trigonostemine A, trigonostemine B and a new synthesis of pityriacitrin and hyrtiosulawesine. Tetrahedron Lett. 2019, 60, 1471–1475. [Google Scholar] [CrossRef]
- Ambule, M.D.; Tripathi, S.; Ghoshal, A.; Srivastava, A.K. IBX-mediated oxidative addition of isocyanides to cyclic secondary amines: Total syntheses of alangiobussine and alangiobussinine. Chem. Commun. 2019, 55, 10872–10875. [Google Scholar] [CrossRef] [PubMed]
- Singha Roy, M.; Meng, X.; Koda, K.; Rasapalli, S.; Gout, D.; Lovely, C.J. Total synthesis of (−)-haploscleridamine. Tetrahedron Lett. 2019, 60, 979–982. [Google Scholar] [CrossRef]
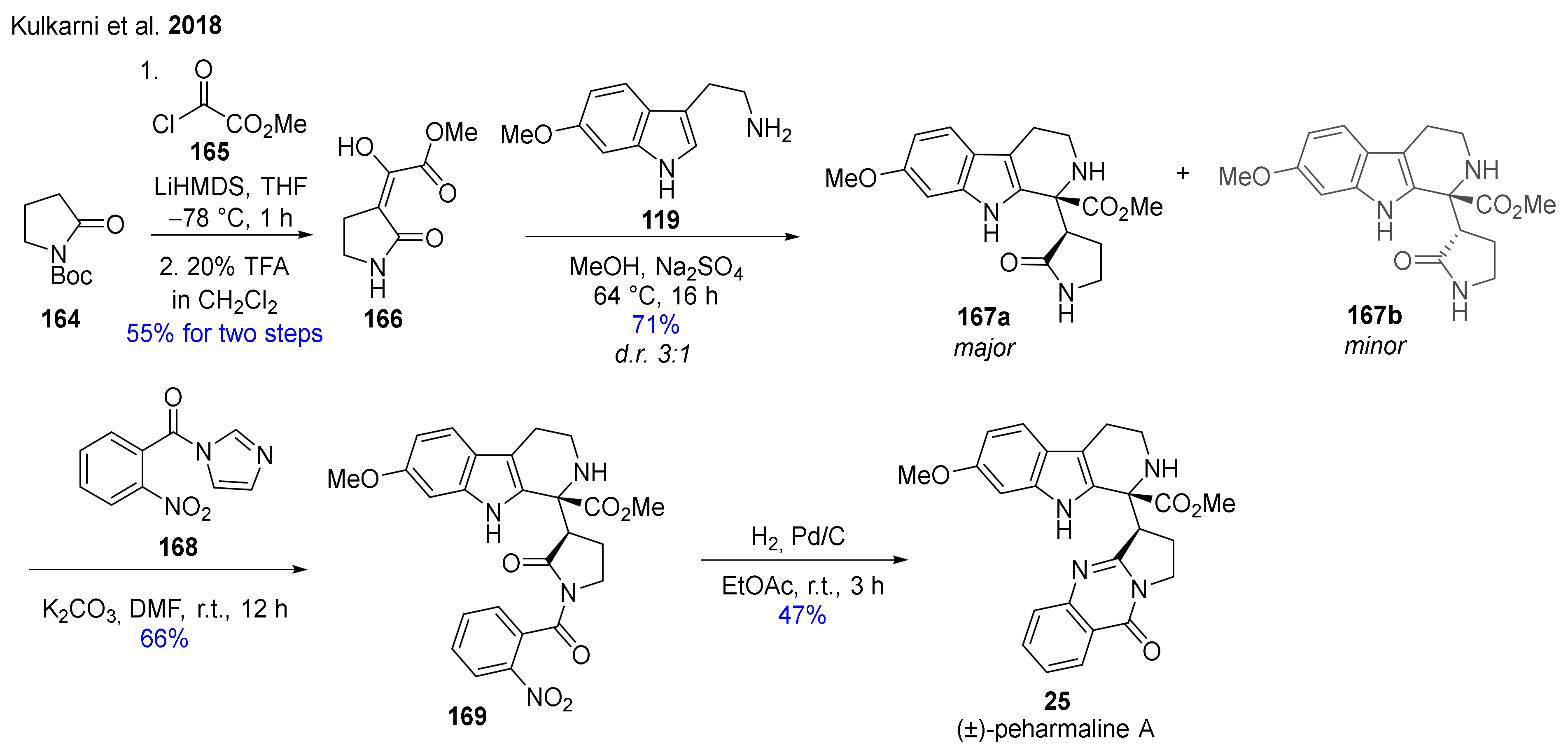
- Kulkarni, A.S.; Shingare, R.D.; Dandela, R.; Reddy, D.S. Total synthesis of an anticancer natural product (±)-peharmaline A and its analogues. Eur. J. Org. Chem. 2018, 2018, 6453–6456. [Google Scholar] [CrossRef]
- Wang, Z.; Niu, J.; Zeng, H.; Li, C.-J. Construction of spirocyclic tetrahydro-β-carbolines via cross-annulation of phenols with tryptamines in water. Org. Lett. 2019, 21, 7033–7037. [Google Scholar] [CrossRef]
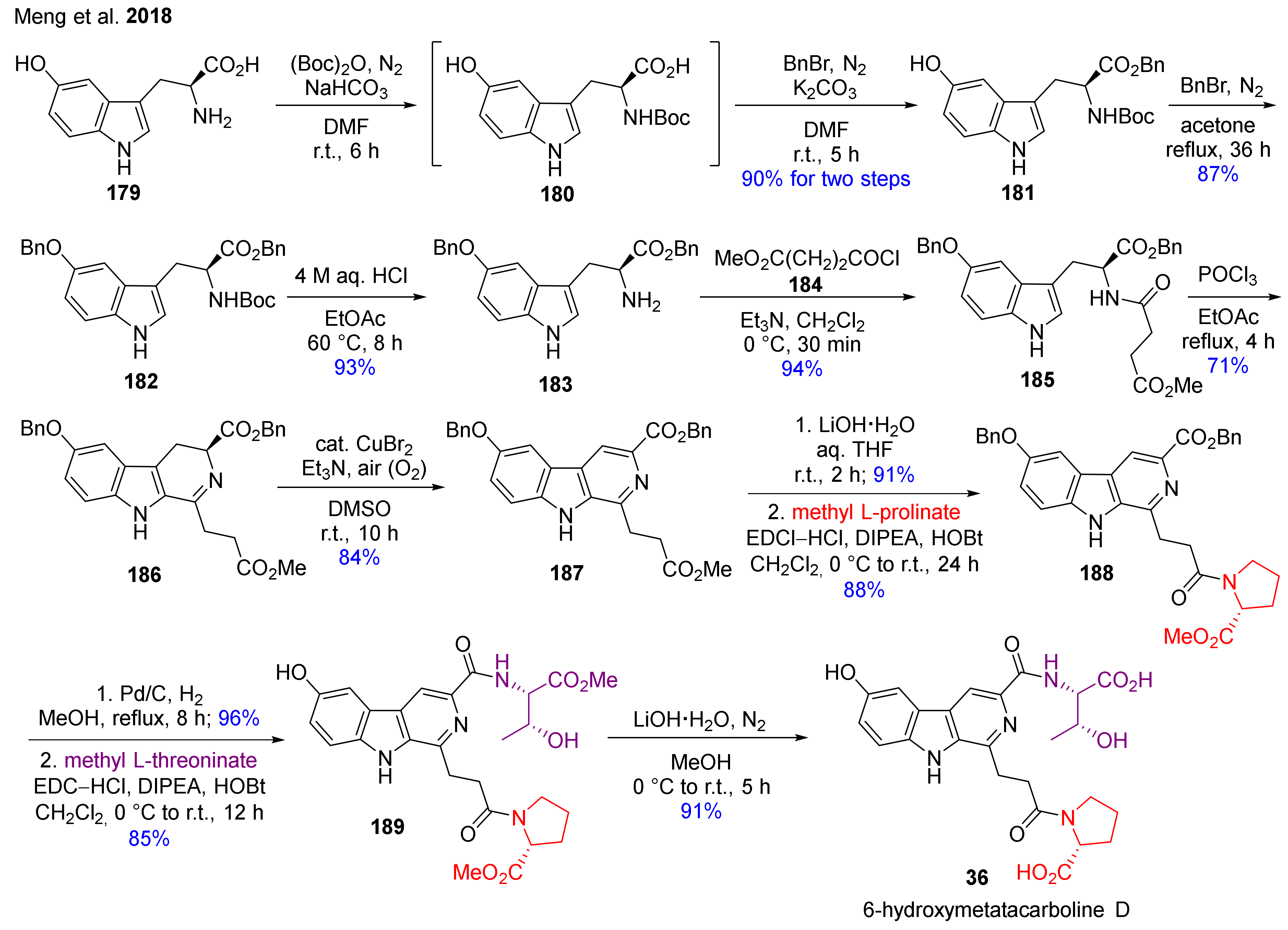
- Meng, T.-Z.; Zheng, J.; Trieu, T.H.; Zheng, B.; Wu, J.-J.; Zhang, Y.; Shi, X.-X. CuBr2-Catalyzed mild oxidation of 3,4-dihydro-β-carbolines and application in total synthesis of 6-hydroxymetatacarboline D. ACS Omega 2018, 3, 544–553. [Google Scholar] [CrossRef] [Green Version]
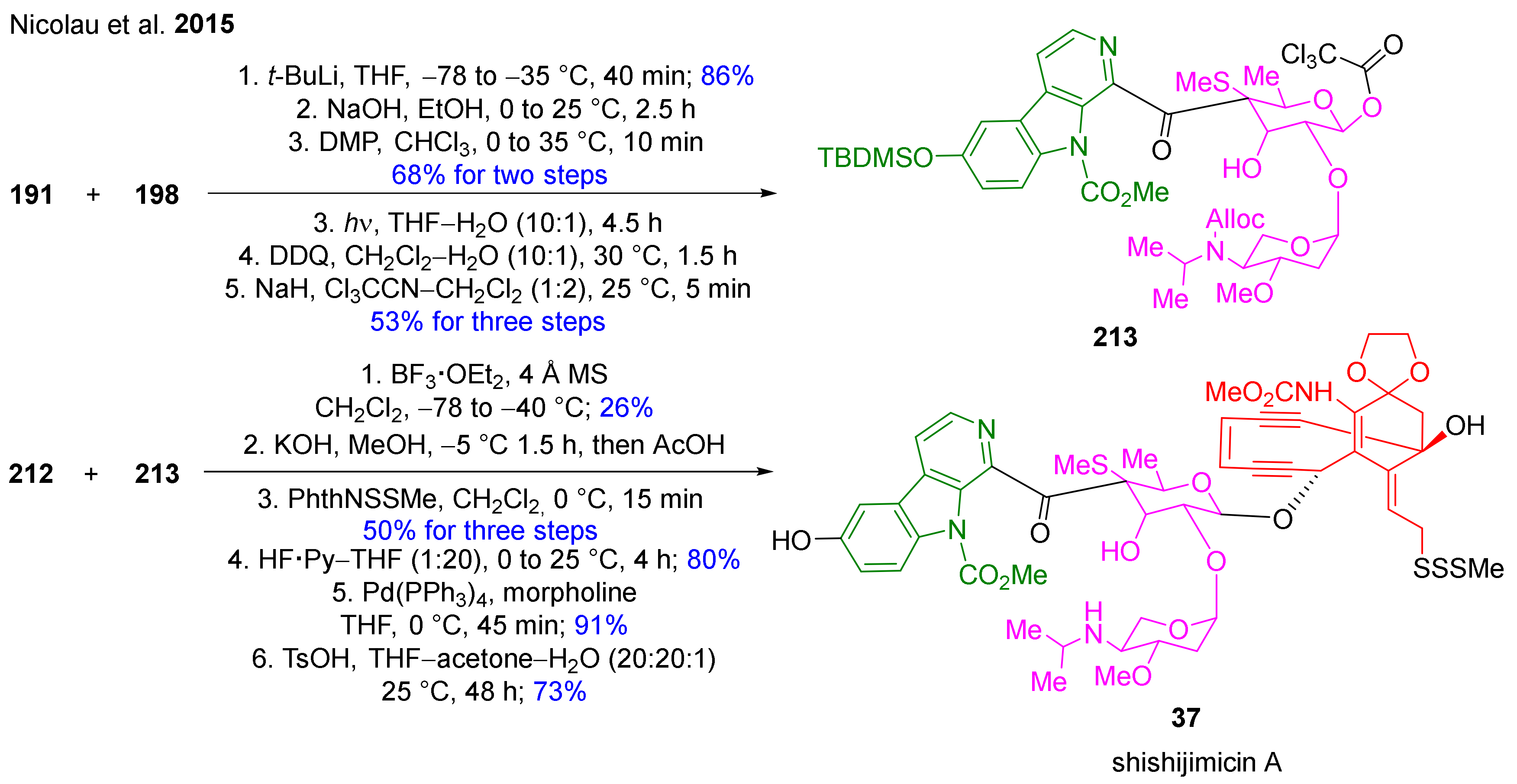
- Nicolaou, K.; Lu, Z.; Li, R.; Woods, J.R.; Sohn, T.-i. Total synthesis of shishijimicin A. J. Am. Chem. Soc. 2015, 137, 8716–8719. [Google Scholar] [CrossRef]
- Schott, Y.; Decker, M.; Rommelspacher, H.; Lehmann, J. 6-Hydroxy- and 6-methoxy-β-carbolines as acetyl- and butyrylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 5840–5843. [Google Scholar] [CrossRef]
- Cogan, D.A.; Liu, G.; Ellman, J. Asymmetric synthesis of chiral amines by highly diastereoselective 1,2-additions of organometallic reagents to N-tert-butanesulfinyl imines. Tetrahedron 1999, 55, 8883–8904. [Google Scholar] [CrossRef]
- Davis, F.A.; Gaddiraju, N.V.; Theddu, N.; Hummel, J.R.; Kondaveeti, S.K.; Zdilla, M.J. Enantioselective synthesis of cocaine C-1 analogues using sulfinimines (N-sulfinyl imines). J. Org. Chem. 2012, 77, 2345–2359. [Google Scholar] [CrossRef]
- Nishikawa, Y.; Kitajima, M.; Kogure, N.; Takayama, H. A divergent approach for the total syntheses of cernuane-type and quinolizidine-type Lycopodium alkaloids. Tetrahedron 2009, 65, 1608–1617. [Google Scholar] [CrossRef]
- Fandrick, D.R.; Hart, C.A.; Okafor, I.S.; Mercadante, M.A.; Sanyal, S.; Masters, J.T.; Sarvestani, M.; Fandrick, K.R.; Stockdill, J.L.; Grinberg, N. Copper-catalyzed asymmetric propargylation of cyclic aldimines. Org. Lett. 2016, 18, 6192–6195. [Google Scholar] [CrossRef] [PubMed]
- Nalikezhathu, A.; Cherepakhin, V.; Williams, T.J. Ruthenium Catalyzed Tandem Pictet–Spengler Reaction. Org. Lett. 2020, 22, 4979–4984. [Google Scholar] [CrossRef] [PubMed]
- Shelar, S.V.; Argade, N.P. Total synthesis of bioactive canthine alkaloid cordatanine comprising in situ double oxidative aromatization of tetrahydrocarbazole. ACS Omega 2017, 2, 3945–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, H.W.; Liao, Y.-R.; Hwang, T.-L.; Shieh, P.-C.; Lee, K.-H.; Hung, H.-Y.; Wu, T.-S. Total synthesis of cordatanine, structural reassignment of drymaritin, and anti-inflammatory activity of synthetic precursors. Bioorg. Med. Chem. Lett. 2015, 25, 3822–3824. [Google Scholar] [CrossRef]
- Zheng, Y.; Wei, K.; Yang, Y.-R. Total synthesis of (−)-geissoschizol through Ir-catalyzed allylic amidation as the key step. Org. Lett. 2017, 19, 6460–6462. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Xu, Z.; Tan, R.; Lei, X. Divergent total synthesis of chaetoglines C to F. J. Org. Chem. 2019, 84, 8766–8770. [Google Scholar] [CrossRef]
- Zhidkov, M.E.; Kantemirov, A.V.; Koisevnikov, A.V.; Andin, A.N.; Kuzmich, A.S. Syntheses of the marine alkaloids 6-oxofascaplysin, fascaplysin and their derivatives. Tetrahedron Lett. 2018, 59, 708–711. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Xiang, J.-C.; Wang, M.; Ma, J.-T.; Wu, Y.-D.; Wu, A.-X. One-Pot Total Synthesis of evodiamine and its analogues through a continuous biscyclization reaction. Org. Lett. 2018, 20, 6380–6383. [Google Scholar] [CrossRef]
- Zhao, Z.; Wei, H.; Xiao, K.; Cheng, B.; Zhai, H.; Li, Y. Facile Synthesis of pyridines from propargyl amines: Concise total synthesis of suaveoline alkaloids. Angew. Chem. 2019, 131, 1160–1164. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Bi, Y.-Z.; Yu, F.-X.; Menzia, G.; Cook, J. Stereospecificity in the Pictet–Spengler reaction. Enantiospecific synthesis of (6S,10S)--(–)-5-methyl-9-oxo-12-benzyl-6,7,8,9,10,11-hexahydro-6,10-imino-5H-cyclooct[b]indole, a template for preparation of macroline/sarpagine alkaloids. Heterocycles 1992, 34, 517–547. [Google Scholar]
- Fu, X.; Cook, J.M. General approach for the synthesis of ajmaline-related alkaloids. Enantiospecific total synthesis of (-)-suaveoline, (-)-raumacline, and (-)-Nb-methylraumacline. J. Org. Chem. 1993, 58, 661–672. [Google Scholar] [CrossRef]
- Smith, M.W.; Zhou, Z.; Gao, A.X.; Shimbayashi, T.; Snyder, S.A. A 7-step formal asymmetric total synthesis of strictamine via an asymmetric propargylation and metal-mediated cyclization. Org. Lett. 2017, 19, 1004–1007. [Google Scholar] [CrossRef]
- Ren, W.; Wang, Q.; Zhu, J. Total synthesis of (±)-strictamine. Angew. Chem. Int. Ed. 2016, 55, 3500–3503. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, D.; Ohara, A.; Chiba, H.; Kumagai, H.; Oishi, S.; Fujii, N.; Ohno, H. Formal total synthesis of (±)-strictamine based on a gold-catalyzed cyclization. Org. Lett. 2016, 18, 1670–1673. [Google Scholar] [CrossRef] [Green Version]
- Kolundžić, F.; Murali, A.; Pérez-Galán, P.; Bauer, J.O.; Strohmann, C.; Kumar, K.; Waldmann, H. A cyclization–rearrangement cascade for the synthesis of structurally complex chiral gold(I)–aminocarbene complexes. Angew. Chem. Int. Ed. 2014, 53, 8122–8126. [Google Scholar] [CrossRef]
- Zheng, Y.; Yue, B.-B.; Wei, K.; Yang, Y.-R. Short synthesis of the monoterpene indole alkaloid (±)-arbornamine. J. Org. Chem. 2018, 83, 4867–4870. [Google Scholar] [CrossRef]
- Szántay, C.N. The Chemistry of Heterocyclic Compounds; Wiley: Chichester, UK, 1994; Volume 25, Chapter 9. [Google Scholar]
- Vas, Á.; Gulyás, B. Eburnamine derivatives and the brain. Med. Res. Rev. 2005, 25, 737–757. [Google Scholar] [CrossRef]
- Massiot, G.; Oliveira, F.; Lévy, J. Synthesis in the indole series. 9. Total synthesis of the pseudovincamones. Bull. Soc. Chim. Fr. Partie 2 1982, 185–190. [Google Scholar]
- Smith, M.W.; Ferreira, J.; Hunter, R.; Venter, G.A.; Su, H. Synthesis of (+)-tacamonine via stereoselective radical cyclization. Org. Lett. 2019, 21, 8740–8745. [Google Scholar] [CrossRef]
- Jarret, M.; Tap, A.; Kouklovsky, C.; Poupon, E.; Evanno, L.; Vincent, G. Bioinspired oxidative cyclization of the geissoschizine skeleton for the total synthesis of (−)-17-nor-excelsinidine. Angew. Chem. Int. Ed. 2018, 57, 12294–12298. [Google Scholar] [CrossRef] [PubMed]
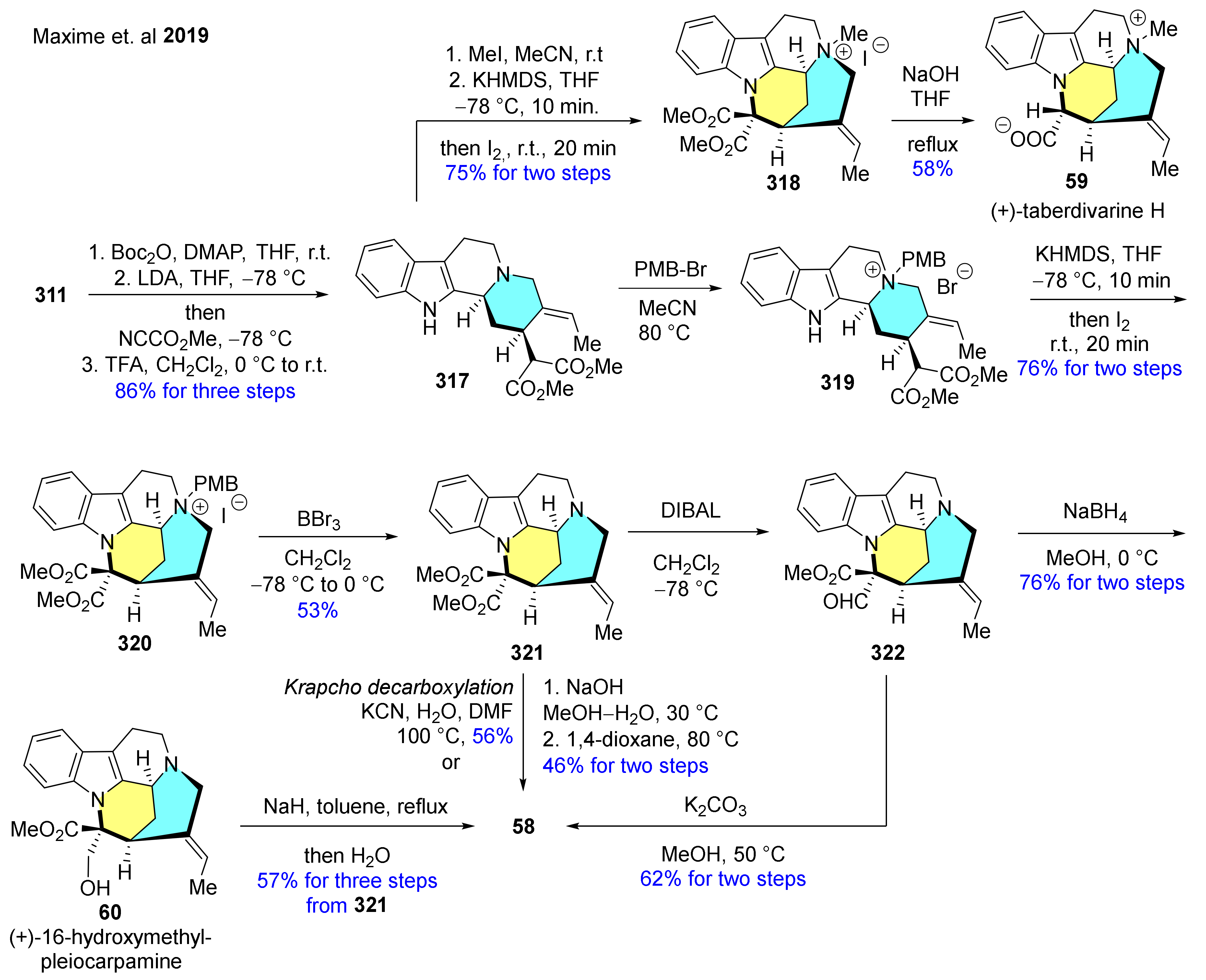
- Maxime, J.; Victor, T.; Aurélien, T.; Jean-Francois, G.; Cyrille, K.; Erwan, P.; Guillaume, V.; Laurent, E. Bioinspired oxidative cyclization of the geissoschizine skeleton for the enantioselective total synthesis of mavacuran alkaloids. Angew. Chem. Int. Ed. 2019, 58, 9861–9865. [Google Scholar] [CrossRef]
- Sato, K.; Kogure, N.; Kitajima, M.; Takayama, H. Total syntheses of pleiocarpamine, normavacurine, and C-mavacurine. Org. Lett. 2019, 21, 3342–3345. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Bai, Y.; Bai, W.-J.; Schultz, J.E. Enantioselective divergent synthesis of C19-oxo eburnane alkaloids via palladium-catalyzed asymmetric allylic alkylation of an N-alkyl-α,β-unsaturated lactam. J. Am. Chem. Soc. 2019, 141, 4811–4814. [Google Scholar] [CrossRef]
- Baran, P.S.; Guerrero, C.A.; Corey, E. Short, enantioselective total synthesis of okaramine N. J. Am. Chem. Soc. 2003, 125, 5628–5629. [Google Scholar] [CrossRef]
- White, K.L.; Mewald, M.; Movassaghi, M. Direct observation of intermediates involved in the interruption of the Bischler–Napieralski reaction. J. Org. Chem. 2015, 80, 7403–7411. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Anderson, J.C. A divergent synthetic route to the vallesamidine and schizozygine alkaloids: Total synthesis of (+)-vallesamidine and (+)-14,15-dehydrostrempeliopine. Angew. Chem. Int. Ed. 2019, 58, 18040–18045. [Google Scholar] [CrossRef]
- Evans, D.A.; Seidel, D. Ni(II)−Bis[(R,R)-N,N‘-dibenzylcyclohexane-1,2-diamine]Br2 catalyzed enantioselective Michael additions of 1,3-dicarbonyl compounds to conjugated nitroalkenes. J. Am. Chem. Soc. 2005, 127, 9958–9959. [Google Scholar] [CrossRef]
- Piemontesi, C.; Wang, Q.; Zhu, J. Enantioselective synthesis of (+)-peganumine A. J. Am. Chem. Soc. 2016, 138, 11148–11151. [Google Scholar] [CrossRef]
- Woodward, R.; Bader, F.; Bickel, H.; Frey, A.; Kierstead, R. The total synthesis of reserpine. Tetrahedron 1958, 2, 1–57. [Google Scholar] [CrossRef]
- Khan, Z.A.; Shahzad, S.A.; Anjum, A.; Bale, A.T.; Naqvi, S.A.R. Synthetic approaches toward the reserpine. Synth. Commun. 2018, 48, 1128–1147. [Google Scholar] [CrossRef]
- Park, J.; Chen, D.Y. A desymmetrization-based total synthesis of reserpine. Angew. Chem. Int. Ed. Engl. 2018, 57, 16152–16156. [Google Scholar] [CrossRef] [PubMed]
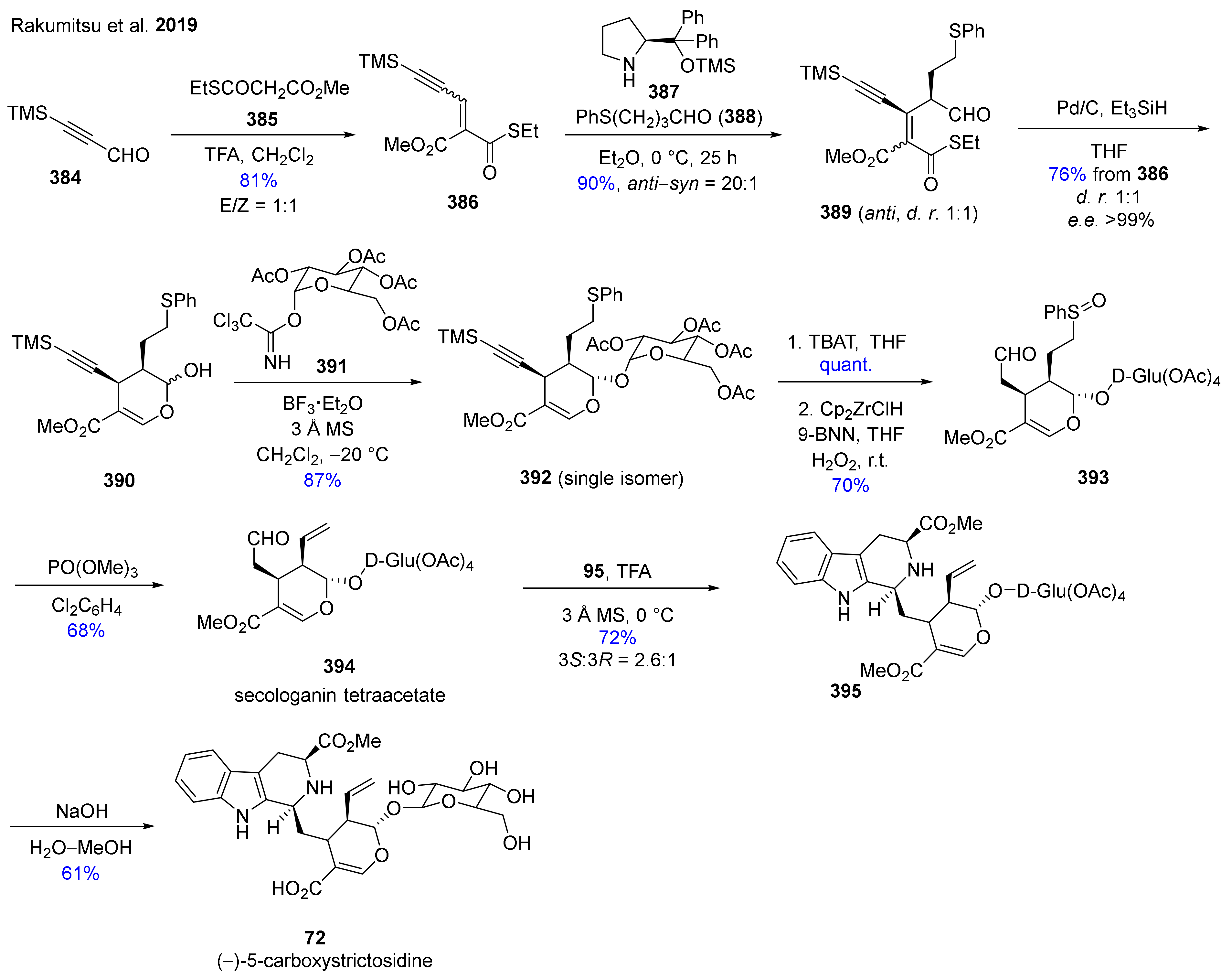
- Rakumitsu, K.; Sakamoto, J.; Ishikawa, H. Total Syntheses of (−)-Secologanin,(−)-5-Carboxystrictosidine, and (−)-Rubenine. Chem. A Eur. J. 2019, 25, 8996–9000. [Google Scholar] [CrossRef] [PubMed]
- Takayama, H.; Fujiwara, R.; Kasai, Y.; Kitajima, M.; Aimi, N. First asymmetric total synthesis of Us-7 and -8, novel D-seco corynanthe-type oxindole alkaloids from Uncaria a ttenuata: Structure revision of Us-7 and determination of absolute stereochemistry. Org. Lett. 2003, 5, 2967–2970. [Google Scholar] [CrossRef]
- Tietze, L.F.; Meier, H.; Nutt, H. Inter- and intramolecular hetero Diels-Alder reactions, 28. Synthesis of (±)-secologanin aglucone O-ethyl ether and derivatives by tandem Knoevenagel hetero Diels-Alder reaction. Liebigs Ann. Chem. 1990, 1990, 253–260. [Google Scholar] [CrossRef]
- Bailey, P.D.; Hollinshead, S.P.; McLay, N.R. Exceptional stereochemical control in the Pictet–Spengler reaction. Tetrahedron Lett. 1987, 28, 5177–5180. [Google Scholar] [CrossRef]


















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Name | Structure | Occurrence | Bioactivity |
|---|---|---|---|---|
| 1 | norharman(β-carboline) |  | Noctiluca milaris [10] Strychnos johnsonii [11] Lycoris longituba [12] Selaginella delicatula [13] Trichosanthes kirilowii Maxim [14] Rauwolfia caffra [15] Didemnum sp. [16] | MAO-B inhibitor11] |
| 2 3 4 5 6 | (−)-tetrahydroharman/ (S)-eleagnine (−)-komaroidine N-(+)-methyltetrahydro-harman (+)-1-ethyl-9-methyl-tetrahydro-β-carboline (+)-N-acetyl-komarodine |  | 2: Albizia polyphylla [17] 3, 6: N. komarovii and N.schoberi [18] 4: Petalostyles labicheoides [19] 5: analogues of natural products | not reported |
| 7 | stolonine C |  | Cnemidocarpa stolonifera [20] | apoptosis in PC3 human prostate cancer cell line [20] |
| 8 9 10 11 12 | eudistomin I eudistomin N eudistomin U eudistomin T isoeudistomin U |  | 8:Eudistoma olivaceum [21] 9: Eudistoma olivaceum [22] 10: Lissoclinum fragile [23] 11: Eudistoma olivaceum [24] 12: analogues of natural products | 8: antibacterial effect [25] 9: antiviral, antibacterial and antifungal activity [26] 11: antibiotic activity [27] |
| 13 14 15 16 | harmalan isoeudistomin M harman kumujian C |  | 13: soybean [28] Flindersia laevicarpa [29] 14: analogues of alcaloids 15: Flindersia Caevicarpa [29] 16: Picrasma quassioides [30] | 13: anti-neuroin-flammatory activities [28] 14: n.a. 15: vasorelaxant effect [31] binding affinity to imidazoline I2B receptors [32] 16: cytotoxic activity [33] AMP phosphodiesterase inhibitor [34] anti-inflammatory agent [35,36] |
| 17 18 19 20 21 | trigonostemine A trigonostemine B trigonostemine G pityriacitrin hyrtiosulawesine |  | 17, 18: Trigonostemone lii [37] 17, 18, 19: Trigonostemone filipes [38] 20: Malassezia furfur [39] Paracoccus marine bacteria [40] Dichotomomyces cejpii [41] 21: Hyrtio erectus [42] Alocasia macrorrhiza [43] | 17, 18, 19: cytotoxic activity [44] 20: antiproliferative activity [45] anticancer activity [46,47] 21: antiproliferative activity [48,49] antimalarial activity [46] antiphospholipase A2 [50] and antioxidant activity [46] |
| 22 23 | alangiobussine alangiobussinine |  | Alangium bussyanum [51] | not reported |
| 24 | haploscleridamine |  | Sponge of the order Haplosclerida [52] | inhibitor of cathepsin K [52] |
| 25 | (±)-peharmaline A |  | Peganum harmala [53] | cytotoxic activity (HL-60, PC-3, SGC-7901) [54] |
| 26 | komavine |  | Nitraria komarorii [55] Nitraria schoberi [55] | not reported |
| 27 28 29 30 | marinacarboline A marinacarboline B marinacarboline C marinacarboline D |  | Marinactinospora thermotolerans SCSIO 00652 [56] | antiplasmodial activity [56] cytotoxic activity [57] |
| 31 | metatacarboline A |  | Mycena metata [58] | not reported |
| 32 33 34 35 | metatacarboline C metatacarboline D metatacarboline E metatacarboline F |  | Mycena metata [58] | 32, 34: antiproliferative activity, proapoptotic effect [59] |
| 36 | 6-hxdroxy- metatacarboline D |  | Mycena metata [58] | not reported |
| 37 | shishijimicin A |  | Didemnum proliferum [60] | antitumor activity (P388 leukemia cells) [60] |
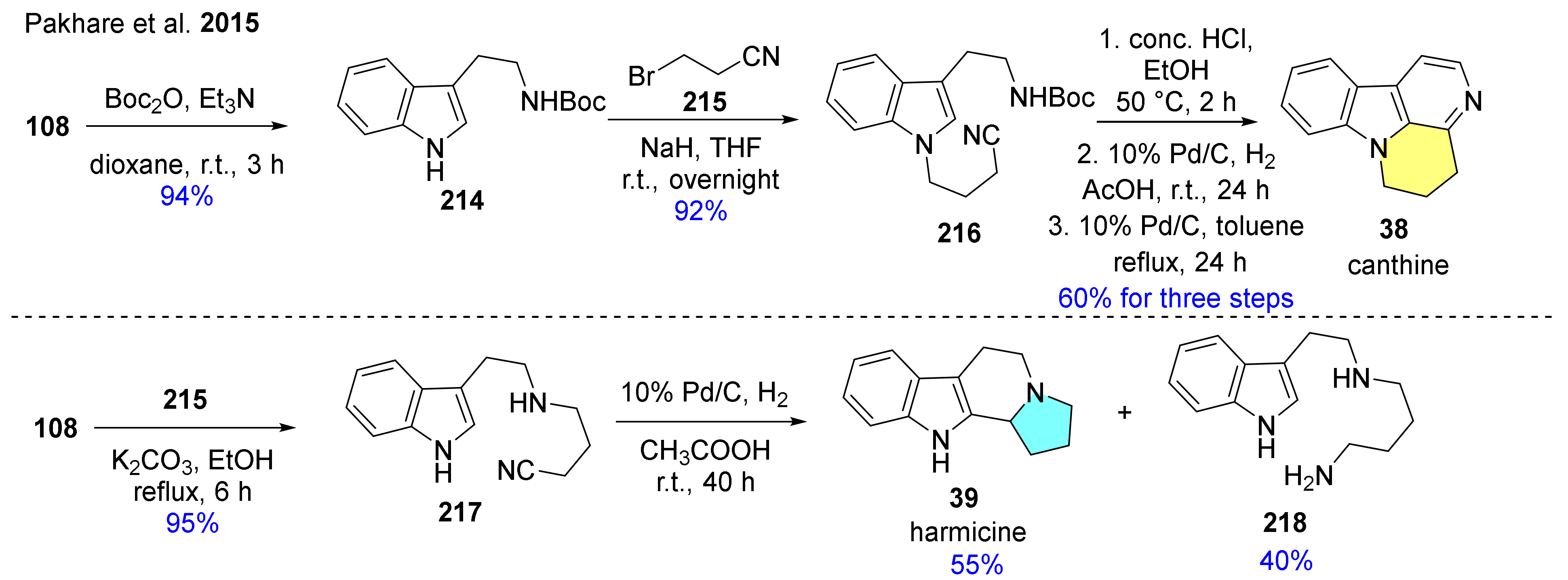
| 38 | canthine |  | basic framework of canthin-6-ones, which are widely distributed in nature [61] | not reported |
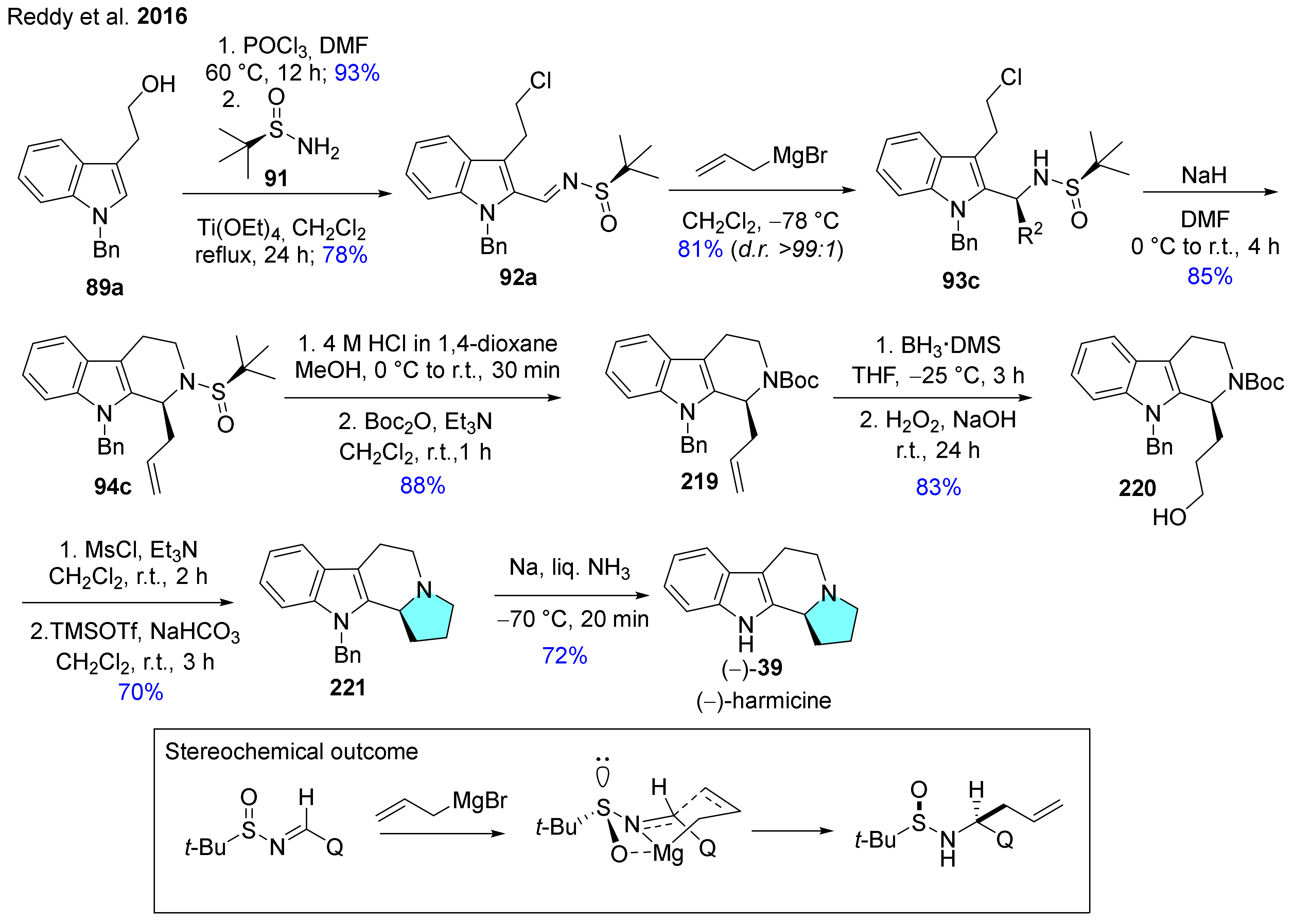
| 39 | harmicine |  | Kopsia griffithii [62] | not reported |
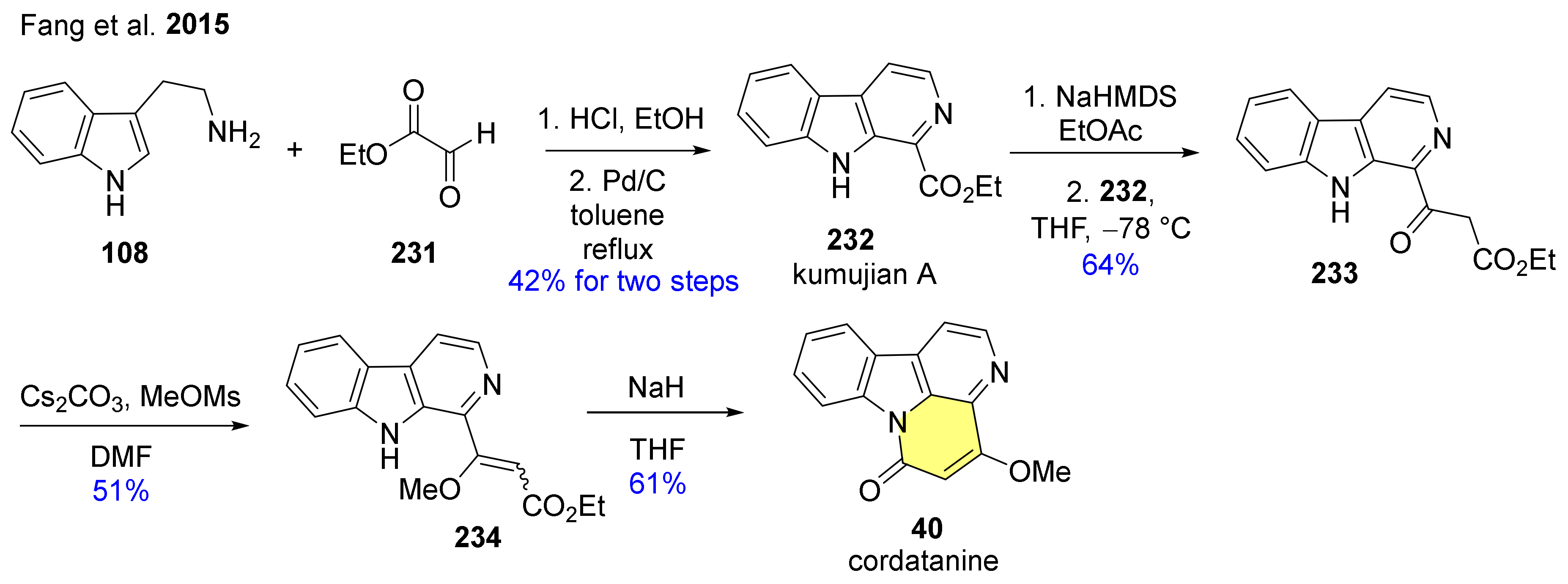
| 40 | cordatanine |  | Drymaria cordata [63] Drymaria diandra [64,65] | anti-HIV activity [64] |
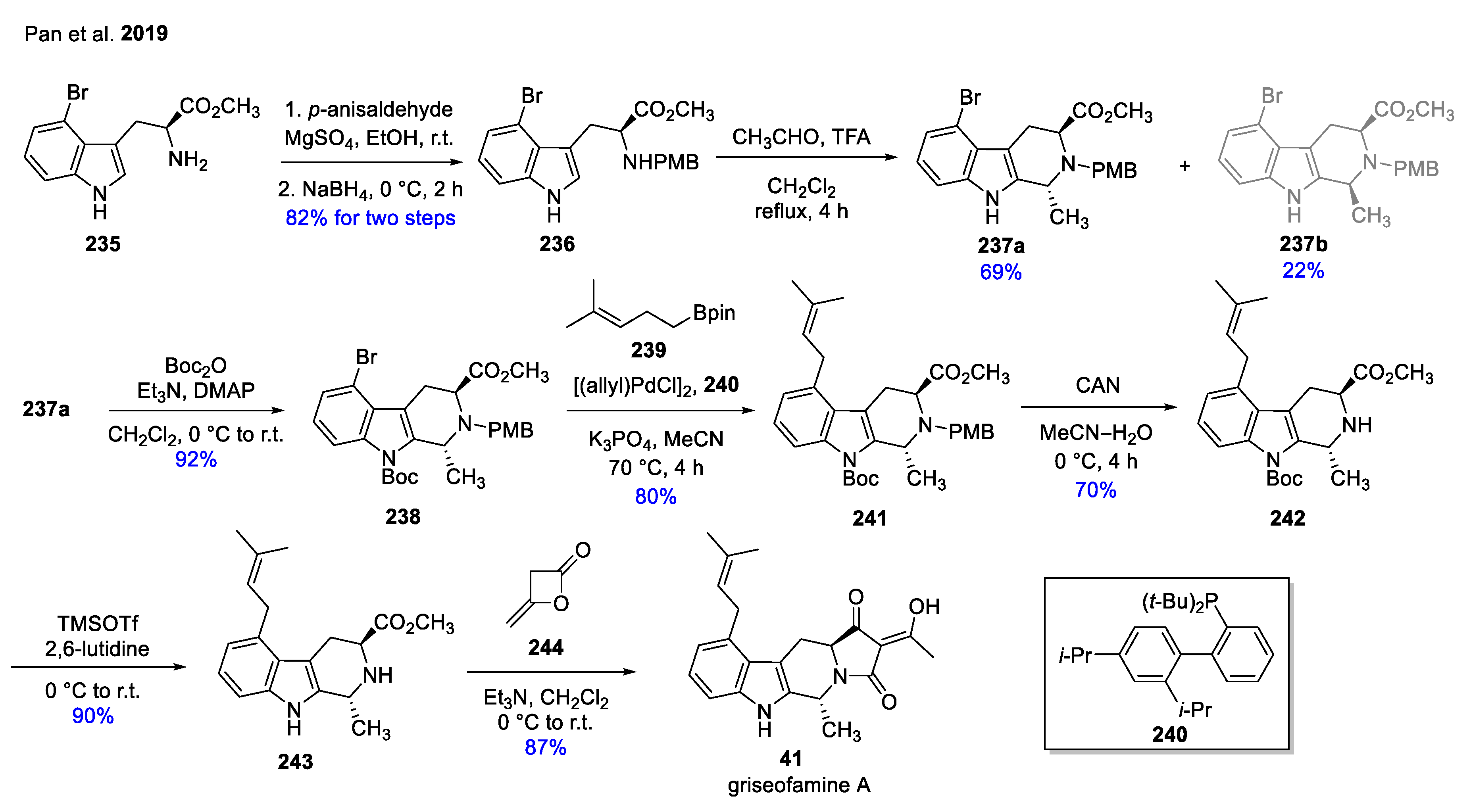
| 41 | griseofamine A |  | Penicillium griseofulvum [66] | antibacterial activity [67] |
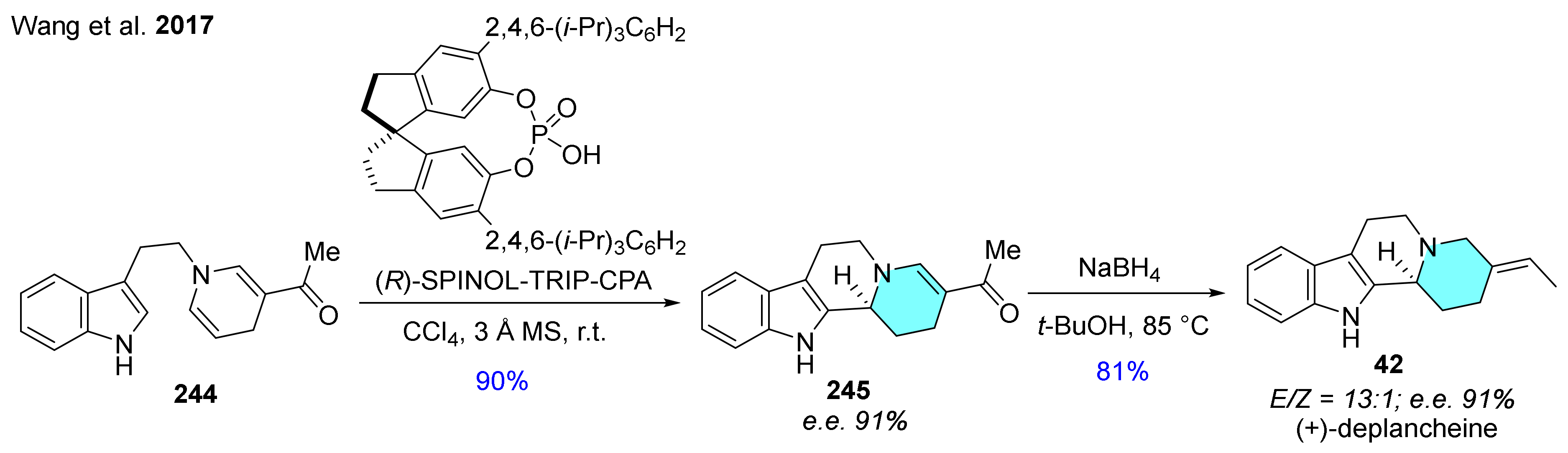
| 42 | (+)-deplancheine |  | Alstonia deplanchei [68] | not reported |
| 43 | (−)-geissoschizol |  | Tabernaemontana bufalina [69] Hunteria zeylanica [70] | not reported |
| 44 | chaetogline E |  | modified Epinephelus drummondhayi with a silent fungal gene from Chaetomium globosum [71] | not reported |
| 45 | chaetogline F |  | modified Epinephelus drummondhayi with a silent fungal gene from Chaetomium globosum [71] | antibacterial activity [71] |
| 46 | 6-oxofascaplysin |  | Hyrtios sp. [72] | weak cytotoxic activity [73] |
| 47 | fascaplysin |  | Fascaplisynopsis sp [74] | anticancer activity [73,75] |
| 48 | evodiamine |  | Evodia rutaecarpa [76] | anticancer anti-inflammatory antimicrobial and many others [77] |
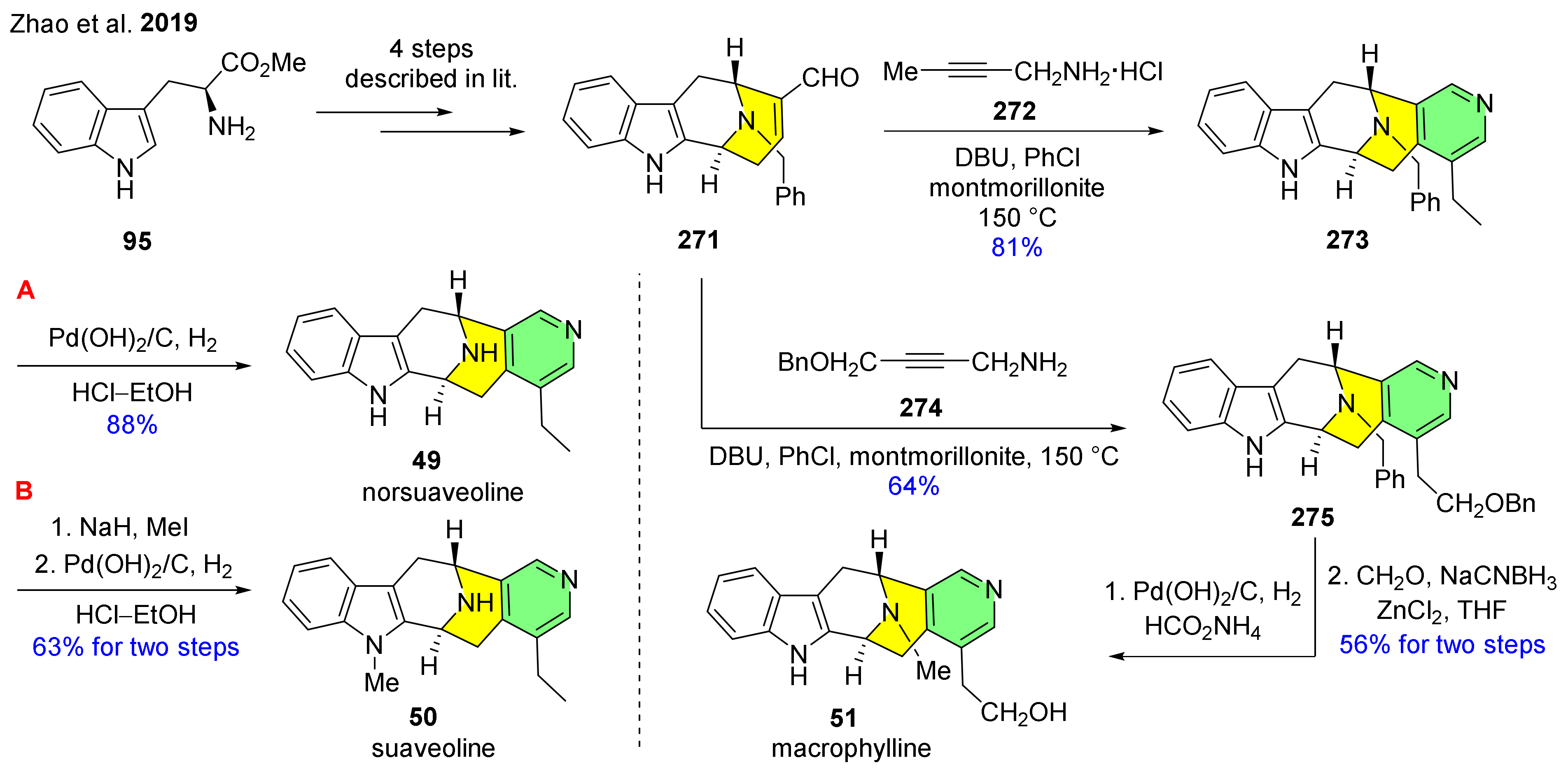
| 49 50 51 | norsuaveoline suaveoline marcophylline |  | 49:Rauvolfia caffra [78] 50: Rauwolfia suaveolens S. Moore (apocynaceae) [79] 51: Rauwolfia macrophylla [80] | 49: not reported 50: acetylcholinesterase inhibitor [81] 51: not reported |
| 52 | (S)-(−)-decarbo-methoxy-dihydro-gambirtannine |  | enantiomer of 52 was isolated from Hunteria zeylanica [82] | not reported |
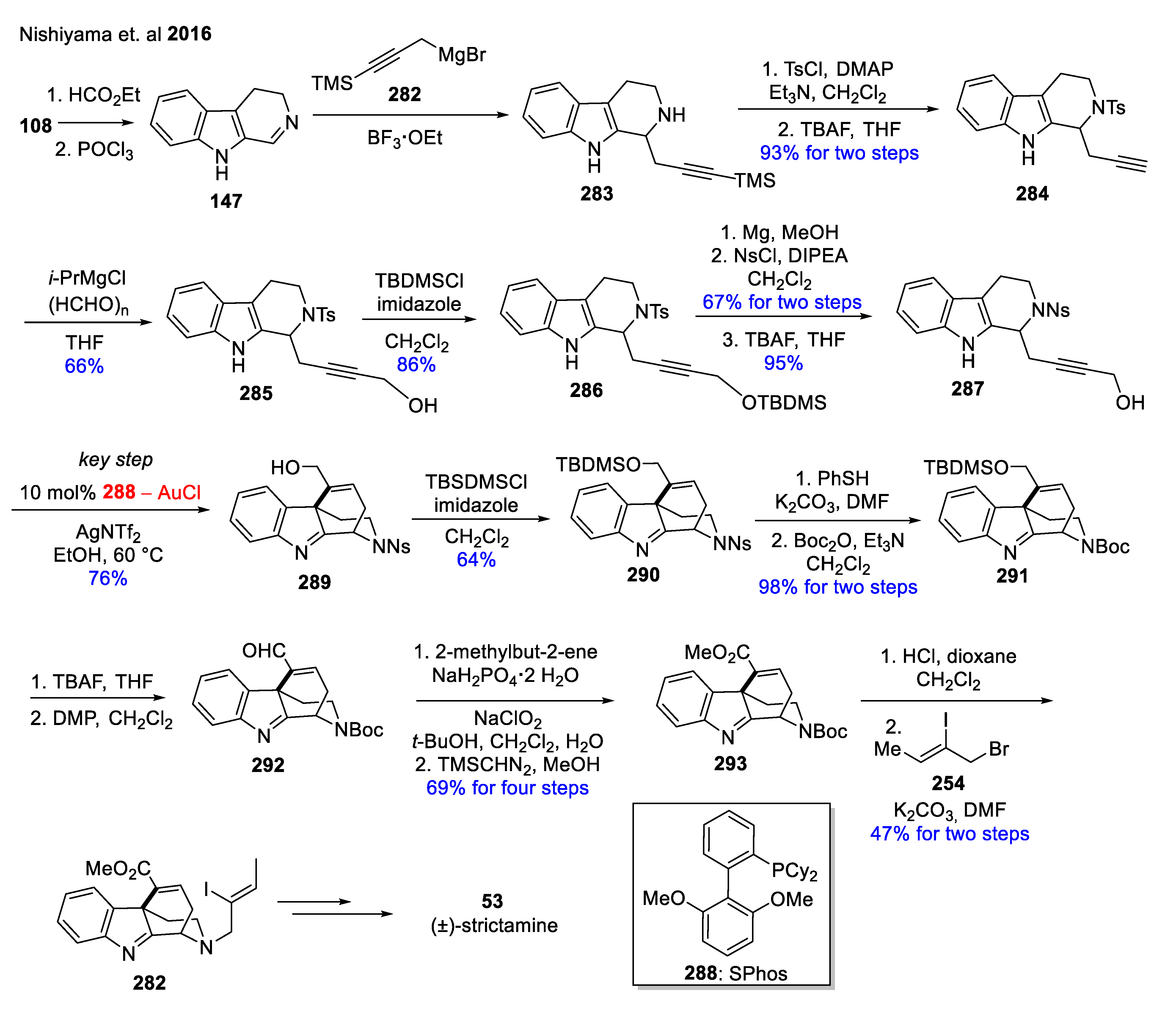
| 53 | (+)-strictamine |  | Alstonis scholaris Rhazya strictu [83,84] | antitumor, antiviral anti-inflammatory antibiosis activities [83,84,85] |
| 54 | (±)-arbornamine |  | Kopsia arborea [86] | not reported |
| 55 | (+)-tacamonine |  | Tabernaemontana eglandulosa [87] | not reported |
| 56 | (−)-17-nor-excelisidine |  | Alstonia scholaris [85] | antiviral activity [85] |
| 57 | (+)-geissoschizine |  | Rhazya stricta [88] | not reported |
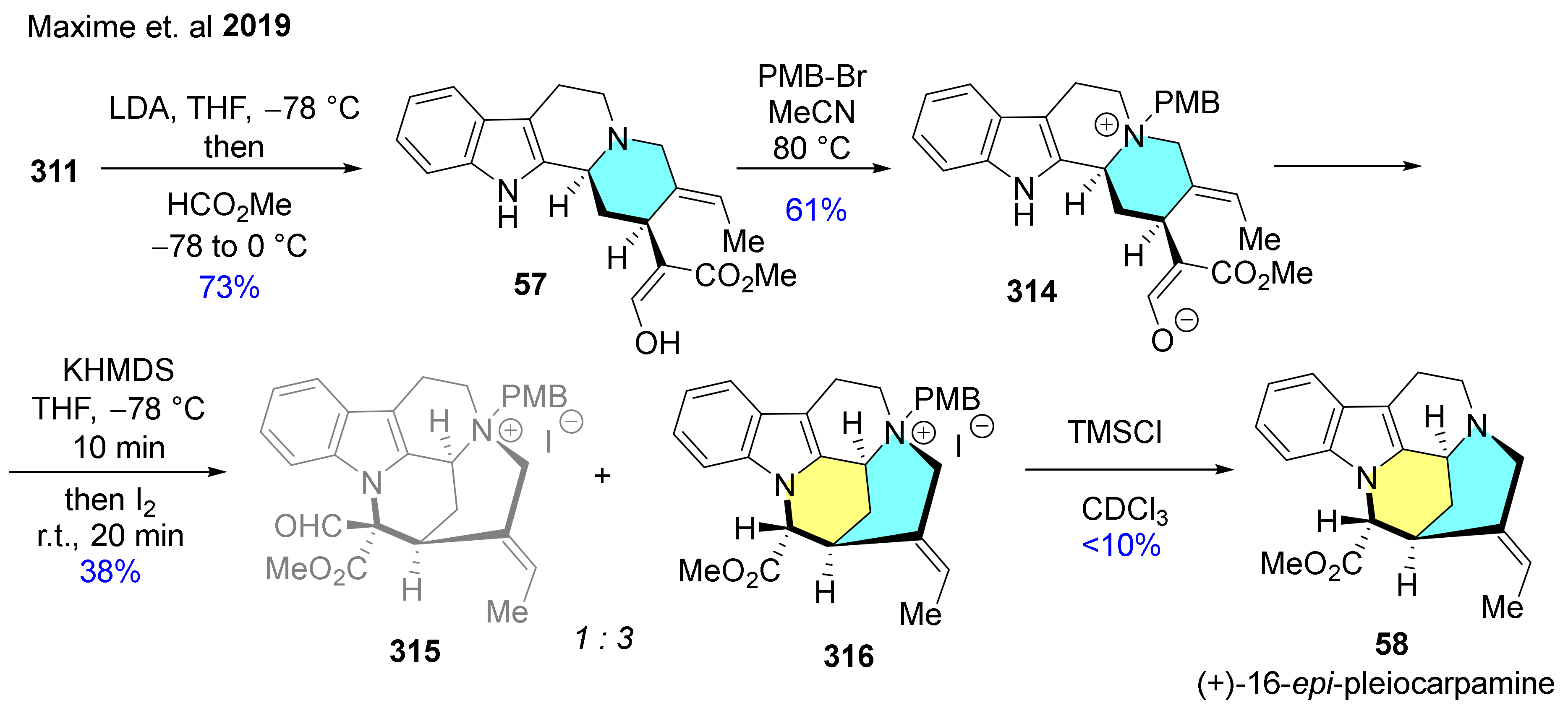
| 58 59 60 61 62 63 | (+)-16-epi-pleiocarpamine (+)-taberdivarine H (+)-16-hydroxymethyl-pleiocarpamine (+)-pleiocarpamine (±)-normavacurine (±)-C-mavacurine |  | 58: Ervatamia cumingiana [89] Pleiocarpa pycnantha [90] 59: Tabernaemontana officinalis [91] 60: Kopsia singapurensis [92] Kopsia teoi [93] 62: Alstonia scholaris [94] 63: Strychnos guianensis [95] | 61, 58: anticholinergic activity [96] 59: cytotoxic activity [91] 60: not reported 62: antibacterial activity [94] 63: not reported |
| 64 65 66 67 | (+)-19-oxoeburnamine (+)-19-OH-eburnamine 19-(S)-hydroxy-Δ14-vincamone (–)-19-hydroxy-Δ14-eburnamonine |  | Kopsia jasminiflora [97] Kopsia pauciflora [98] | not reported closely related analogues show antitumor activity |
| 68 | (+)-vallesamidine |  | Vallesia dichotoma [99] | not reported |
| 69 | (+)-14,15-dehydro-strempeliopine |  | Schizozygia caffaeoides [100] | not reported |
| 70 | (+)-peganumine A |  | Peganum harmala L. [101] | cytotoxic activity (MCF-70, PC-3, HepG2, HL-60) [101] |
| 71 | reserpine |  | Rauwolfia serpentina [102] | Commercially available drug for the treatment of high blood pressure [103] |
| 72 | (–)-5-carboxystrictosidine |  | Rhazya orientalis [104] Guenttarda platypoda [105] Uncaria tomentosa [106] Ophiorrhiza nutans [107] | not reported |
| 73 | (–)-rubenine |  | Adina rubescens [108] | not reported |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szabó, T.; Volk, B.; Milen, M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules 2021, 26, 663. https://doi.org/10.3390/molecules26030663
Szabó T, Volk B, Milen M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules. 2021; 26(3):663. https://doi.org/10.3390/molecules26030663
Chicago/Turabian StyleSzabó, Tímea, Balázs Volk, and Mátyás Milen. 2021. "Recent Advances in the Synthesis of β-Carboline Alkaloids" Molecules 26, no. 3: 663. https://doi.org/10.3390/molecules26030663
APA StyleSzabó, T., Volk, B., & Milen, M. (2021). Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules, 26(3), 663. https://doi.org/10.3390/molecules26030663