
Advances and Perspectives in the Management of Varicella-Zoster Virus Infections



Abstract
:1. Introduction
2. Clinical Characteristics of VZV Infections
3. Vaccination Strategies and Post-Exposure Prophylaxis
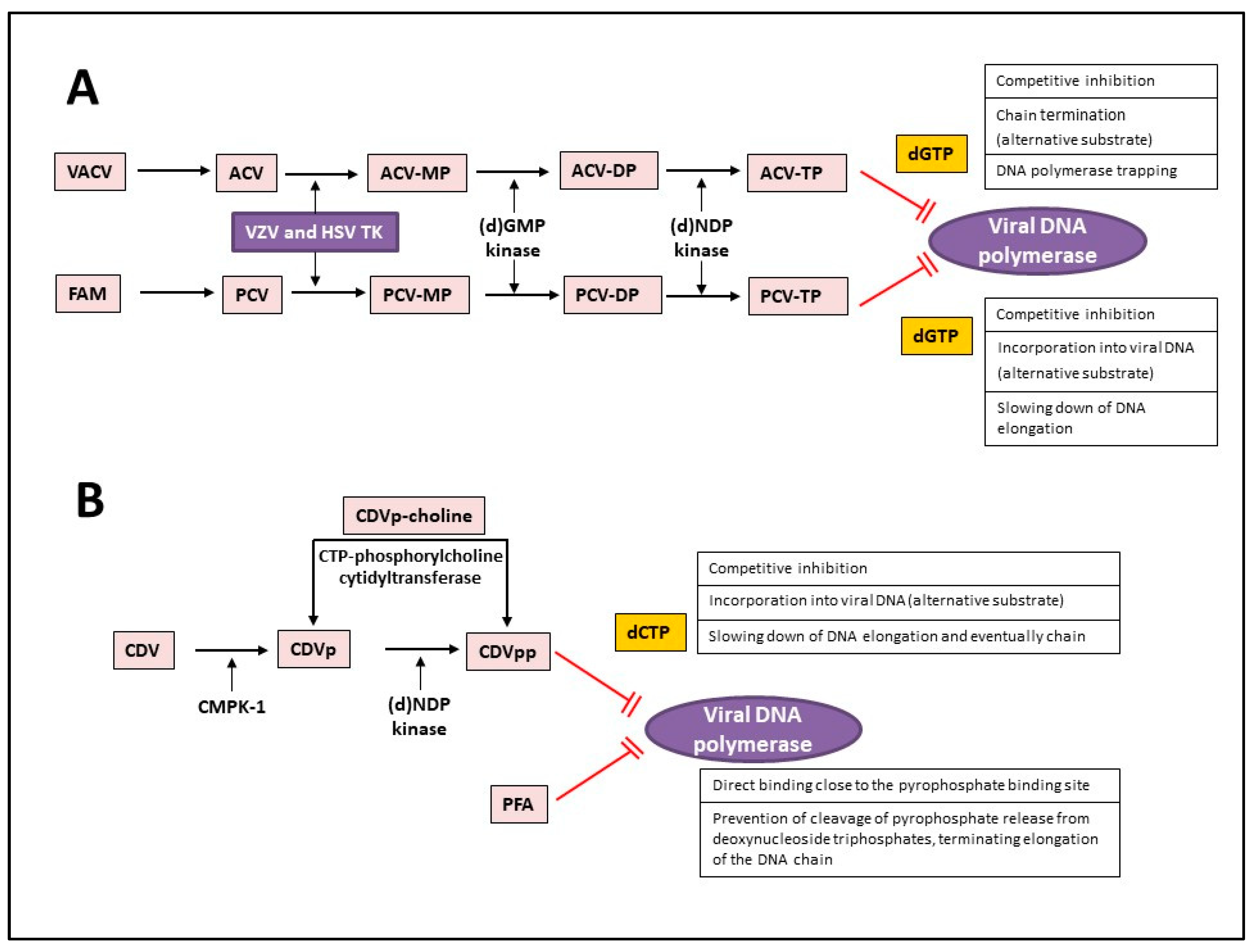
4. Treatment of VZV-Associated Diseases
5. Medical Need for New Antiviral Agents to Manage VZV-Associated Diseases
5.1. Management of PHN and other Complications
5.2. Managing Rare but Significant Side Effects Linked to the VZV Life Vaccine
5.3. Emergence of Drug-Resistant Viruses
6. Novel Anti-VZV Agents in Advanced Development
6.1. Bicyclic Nucleoside Analogues (BCNAs)
6.2. Carbocyclic Nucleoside Analogues: H2G (Omaciclovir) and Its Prodrug (Valomaciclovir)
6.3. Brincidofovir
7. Candidate anti-VZV Drugs
7.1. Nucleoside/Nucleotide Analogues
7.1.1. Phenoxazine Derivatives
7.1.2. 2′-Deoxyribose Emimycin Nucleosides
7.1.3. C5-substituted-(1,3-diyne)-2-deoxyuridines
7.1.4. Carbocyclic Nucleosides: C-3 halo and 3-methyl substituted 5′-nor-3-deazaaristeromycins
7.1.5. Xanthine-Based Acyclic Nucleoside Phosphonates (ANPs)
7.1.6. Cyclopentyl Nucleoside Phosphonates
7.1.7. (E)-but-2-enyl Nucleoside Phosphonoamidates
7.1.8. Prodrugs of C5-Substituted Pyrimidine Acyclic Nucleosides for Antiviral Therapy
7.1.9. Prodrugs of the Pyrimidine Acyclic Nucleoside Phosphonates PMEO-DAPy and PME-5-azaC
7.1.10. Diamyl Aspartate Amidate Prodrugs of 3-Fluoro-2-(phosphonomethoxy)propyl Acyclic Nucleoside Phosphonates
7.1.11. Amidate Prodrugs of Cyclic 9-(S)-[3-Hydroxy-2-(phosphonomethoxy)propyl]adenine, cHPMPA
7.2. Non-Nucleoside Analogues
7.2.1. Pyrazolo[1,5-c]1,3,5-triazin-4-one Derivative
7.2.2. 5-Chlorobenzo[b]thiophen Derivative
7.2.3. Thienylcarboxamide Derivative
7.2.4. Indole-Based Derivatives
7.2.5. Cephalotaxine Esters
7.3. Hybrid Molecules
Dihydropyrimidinone/1,2,3-triazole Hybrid Molecules
8. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Breuer, J.; Whitley, R. Varicella zoster virus: Natural history and current therapies of varicella and herpes zoster. Herpes 2007, 14, 25–29. [Google Scholar]
- Gershon, A.A.; Gershon, M.D.; Breuer, J.; Levin, M.J.; Oaklander, A.L.; Griffiths, P.D. Advances in the understanding of the pathogenesis and epidemiology of herpes zoster. J. Clin. Virol. 2010, 48, S2–S7. [Google Scholar] [CrossRef] [Green Version]
- Defaux, B.A.; Brabant, S.; Chatellier, D.; Bourgoin, A.; Robert, R.; Ruckes, T.; Agius, G. Disseminated varicella with multiorgan failure in an immunocompetent adult. J. Med. Virol. 2009, 81, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Scotch, A.H.; Hoss, E.; Orenstein, R.; Budavari, A.I. Disseminated Varicella-Zoster Virus After Vaccination in an Immunocompetent Patient. J. Am. Osteopath. Assoc. 2016, 116, 402–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrun, B.; Williams, V.; Brice, S. Disseminated varicella-zoster virus in an immunocompetent adult. Dermatol. Online J. 2015, 21. [Google Scholar] [CrossRef] [Green Version]
- Loftus, M.J.; Yong, M.K.; Wilson, S.; Peleg, A.Y. Fatal disseminated visceral varicella zoster virus infection in a renal transplant recipient. Transpl. Infect. Dis. 2019, 21, 3062. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.J.; Schlichte, M.J.; Dao, H., Jr. Atypical disseminated herpes zoster: Management guidelines in immunocompromised patients. Cutis 2017, 100, 321–330. [Google Scholar]
- Johnson, R.W.; McElhaney, J. Postherpetic neuralgia in the elderly. Int. J. Clin. Pract. 2009, 63, 1386–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opstelten, W.; McElhaney, J.; Weinberger, B.; Oaklander, A.L.; Johnson, R.W. The impact of varicella zoster virus: Chronic pain. J. Clin. Virol. 2010, 48, S8–S13. [Google Scholar] [CrossRef]
- McElhaney, J.E. Herpes zoster: A common disease that can have a devastating impact on patients’ quality of life. Expert Rev. Vaccines 2010, 9, 27–30. [Google Scholar] [CrossRef]
- Davis, A.R.; Sheppard, J. Herpes Zoster Ophthalmicus Review and Prevention. Eye Contact Lens. 2019, 45, 286–291. [Google Scholar] [CrossRef]
- Kedar, S.; Jayagopal, L.N.; Berger, J.R. Neurological and Ophthalmological Manifestations of Varicella Zoster Virus. J. Neuroophthalmol. 2019, 39, 220–231. [Google Scholar] [CrossRef]
- Depledge, D.P.; Cudini, J.; Kundu, S.; Atkinson, C.; Brown, J.R.; Haque, T.; Houldcroft, C.J.; Koay, E.S.; McGill, F.; Milne, R.; et al. High Viral Diversity and Mixed Infections in Cerebral Spinal Fluid from Cases of Varicella Zoster Virus Encephalitis. J. Infect. Dis. 2018, 218, 1592–1601. [Google Scholar] [CrossRef] [Green Version]
- Wauters, O.; Lebas, E.; Nikkels, A.F. Chronic mucocutaneous herpes simplex virus and varicella zoster virus infections. J. Am. Acad. Dermatol. 2012, 66, e217–e227. [Google Scholar] [CrossRef]
- Johnson, R.W.; Pasquin, A.M.J.; Bijl, M.; Franco, E.; Gaillat, J.; Clara, J.G.; Labetoulle, M.; Michel, J.P.; Naldi, L.; Sanmarti, L.S.; et al. Herpes zoster epidemiology, management, and disease and economic burden in Europe: A multidisciplinary perspective. Ther. Adv. Vaccines 2015, 3, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, N.H.; Gilden, D.H.; Cohrs, R.J.; Mahalingam, R.; Nagel, M.A. Varicella zoster virus infection: Clinical features, molecular pathogenesis of disease, and latency. Neurol. Clin. 2008, 26, 675–697. [Google Scholar] [CrossRef] [Green Version]
- Bergstrom, R.; Tripathy, K. Acute Retinal Necrosis; StatPearls: Treasure Island, FL, USA, 2019. [Google Scholar]
- Pavesio, C.E.; Mitchell, S.M.; Barton, K.; Schwartz, S.D.; Towler, H.M.; Lightman, S. Progressive outer retinal necrosis (PORN) in AIDS patients: A different appearance of varicella-zoster retinitis. Eye 1995, 9, 271–276. [Google Scholar] [CrossRef] [Green Version]
- Yin, P.D.; Kurup, S.K.; Fischer, S.H.; Rhee, H.H.; Byrnes, G.A.; Clarke, L.G.A.; Buggage, R.R.; Nussenblatt, R.B.; Mican, J.M.; Wright, M.E. Progressive outer retinal necrosis in the era of highly active antiretroviral therapy: Successful management with intravitreal injections and monitoring with quantitative PCR. J. Clin. Virol. 2007, 38, 254–259. [Google Scholar] [CrossRef]
- Austin, R.B. Progressive outer retinal necrosis syndrome: A comprehensive review of its clinical presentation, relationship to immune system status, and management. Clin. Eye Vis. Care 2000, 12, 119–129. [Google Scholar] [CrossRef]
- Gunturu, M.; Gosi, S.K.; Kanduri, S.; Garla, V. Varicella Zoster Meningitis, Optic Neuritis Preceding the Development of Posterior Outer Retinal Necrosis, and Central Retinal Artery Occlusion in a HIV Patient. Case Rep. Med. 2019, 2019, 3162. [Google Scholar] [CrossRef]
- Baxter, R.; Ray, P.; Tran, T.N.; Black, S.; Shinefield, H.R.; Coplan, P.M.; Lewis, E.; Fireman, B.; Saddier, P. Long-term effectiveness of varicella vaccine: A 14-Year, prospective cohort study. Pediatrics 2013, 131, e1389–e1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, M.; Leung, J.; Gershon, A.A. Transmission of Vaccine-Strain Varicella-Zoster Virus: A Systematic Review. Pediatrics 2019, 144. [Google Scholar] [CrossRef]
- Norberg, P.; Depledge, D.P.; Kundu, S.; Atkinson, C.; Brown, J.; Haque, T.; Hussaini, Y.; MacMahon, E.; Molyneaux, P.; Papaevangelou, V.; et al. Recombination of Globally Circulating Varicella-Zoster Virus. J. Virol. 2015, 89, 7133–7146. [Google Scholar] [CrossRef] [Green Version]
- Bastidas, A.; de la Serna, J.; El Idrissi, M.; Oostvogels, L.; Quittet, P.; Jimenez, L.J.; Vural, F.; Pohlreich, D.; Zuckerman, T.; Issa, N.C.; et al. Effect of Recombinant Zoster Vaccine on Incidence of Herpes Zoster After Autologous Stem Cell Transplantation: A Randomized Clinical Trial. JAMA 2019, 322, 123–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, C.M.; Faulds, D. Valaciclovir. A review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy in herpesvirus infections. Drugs 1996, 52, 754–772. [Google Scholar] [CrossRef]
- Perry, C.M.; Wagstaff, A.J. Famciclovir. A review of its pharmacological properties and therapeutic efficacy in herpesvirus infections. Drugs 1995, 50, 396–415. [Google Scholar] [CrossRef] [PubMed]
- Hodge, V.R.A.; Sutton, D.; Boyd, M.R.; Harnden, M.R.; Jarvest, R.L. Selection of an oral prodrug (BRL 42810; famciclovir) for the antiherpesvirus agent BRL 39123 (9-(4-hydroxy-3-hydroxymethylbut-l-yl)guanine; penciclovir). Antimicrob. Agents Chemother. 1989, 33, 1765–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clercq, E. Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster. Biochem. Pharmacol. 2004, 68, 2301–2315. [Google Scholar] [CrossRef]
- Wutzler, P. Antiviral therapy of herpes simplex and varicella-zoster virus infections. Intervirology 1997, 40, 343–356. [Google Scholar] [CrossRef]
- Yaldiz, M.; Solak, B.; Kara, R.O.; Cosansu, N.; Erdem, M.T. Comparison of Famciclovir, Valaciclovir, and Brivudine Treatments in Adult Immunocompetent Patients with Herpes Zoster. Am. J. Ther. 2018, 25, e626–e634. [Google Scholar] [CrossRef]
- Desgranges, C.; Razaka, G.; De Clercq, E.; Herdewijn, P.; Balzarini, J.; Drouillet, F.; Bricaud, H. Effect of (E)-5-(2-bromovinyl)uracil on the catabolism and antitumor activity of 5-fluorouracil in rats and leukemic mice. Cancer Res. 1986, 46, 1094–1101. [Google Scholar]
- Tsifi, A.; Papaxoinis, G.; Diamantopoulos, P.; Mantzourani, M.; Antoniadou, V.; Halioti, A.; Gogas, H. A life-threatening drug-drug interaction between capecitabine and brivudine in a patient with metastatic breast cancer. J. Chemother. 2019, 31, 424–427. [Google Scholar] [CrossRef]
- Canada, B.J.M.; Martinez, M.J.; Olmedo, G.O.; Barcenas, J.R.; Cueto, M.P. Interaction between capecitabine and brivudin in a patient with breast cancer. Nat. Rev. Clin. Oncol. 2010, 7, 55–58. [Google Scholar] [CrossRef]
- Bravo, R.A.E.; Hofer, S.; Krahenbuhl, S.; Ludwig, C. Fatal drug-drug interaction of brivudine and capecitabine. Acta Oncol. 2009, 48, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Ogura, K.; Kato, A.; Takubo, H.; Watabe, T. A possible mechanism of eighteen patient deaths caused by interactions of sorivudine, a new antiviral drug, with oral 5-fluorouracil prodrugs. J. Pharmacol. Exp. Ther. 1998, 287, 791–799. [Google Scholar]
- Okuda, H.; Nishiyama, T.; Ogura, K.; Nagayama, S.; Ikeda, K.; Yamaguchi, S.; Nakamura, Y.; Kawaguchi, K.; Watabe, T.; Ogura, Y. Lethal drug interactions of sorivudine, a new antiviral drug, with oral 5-fluorouracil prodrugs. Drug Metab. Dispos. 1997, 25, 270–273. [Google Scholar] [PubMed]
- Chono, K.; Katsumata, K.; Kontani, T.; Kobayashi, M.; Sudo, K.; Yokota, T.; Konno, K.; Shimizu, Y.; Suzuki, H. ASP2151, a novel helicase-primase inhibitor, possesses antiviral activity against varicella-zoster virus and herpes simplex virus types 1 and 2. J. Antimicrob. Chemother. 2010, 65, 1733–1741. [Google Scholar] [CrossRef] [Green Version]
- Kleymann, G. New antiviral drugs that target herpesvirus helicase primase enzymes. Herpes 2003, 10, 46–52. [Google Scholar]
- Chono, K.; Katsumata, K.; Suzuki, H.; Shiraki, K. Synergistic activity of amenamevir (ASP2151) with nucleoside analogs against herpes simplex virus types 1 and 2 and varicella-zoster virus. Antivir. Res. 2012, 97, 154–160. [Google Scholar] [CrossRef]
- Chono, K.; Katsumata, K.; Kontani, T.; Shiraki, K.; Suzuki, H. Characterization of virus strains resistant to the herpes virus helicase-primase inhibitor ASP2151 (Amenamevir). Biochem. Pharmacol. 2012, 84, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Birkmann, A.; Zimmermann, H. Drugs in development for herpes simplex and varicella zoster virus. Clin. Pharmacol. Ther. 2017, 102, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, M.; Nemoto, O.; Honda, M.; Watanabe, D.; Nakayama, J.; Imafuku, S.; Kato, T.; Katsuramaki, T.; Study, I. Amenamevir, a novel helicase-primase inhibitor, for treatment of herpes zoster: A randomized, double-blind, valaciclovir-controlled phase 3 study. J. Dermatol. 2017, 44, 1219–1227. [Google Scholar] [CrossRef]
- Whitley, R.J.; Volpi, A.; McKendrick, M.; Wijck, A.; Oaklander, A.L. Management of herpes zoster and post-herpetic neuralgia now and in the future. J. Clin. Virol. 2010, 48, S20–S28. [Google Scholar] [CrossRef]
- Levy, O.; Orange, J.S.; Hibberd, P.; Steinberg, S.; La Russa, P.; Weinberg, A.; Wilson, S.B.; Shaulov, A.; Fleisher, G.; Geha, R.S.; et al. Disseminated varicella infection due to the vaccine strain of varicella-zoster virus, in a patient with a novel deficiency in natural killer T cells. J. Infect. Dis. 2003, 188, 948–953. [Google Scholar] [CrossRef] [Green Version]
- Galea, S.A.; Sweet, A.; Beninger, P.; Steinberg, S.P.; Larussa, P.S.; Gershon, A.A.; Sharrar, R.G. The safety profile of varicella vaccine: A 10-year review. J. Infect. Dis. 2008, 197, S165–S169. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Kim, V.; Lavi, S.; Jones, F.E.L.; Tipples, G.; Scolnik, D.; Tellier, R. Vaccine-strain varicella zoster virus causing recurrent herpes zoster in an immunocompetent 2-year-old. Pediatr. Infect. Dis. J. 2008, 27, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Uebe, B.; Sauerbrei, A.; Burdach, S.; Horneff, G. Herpes zoster by reactivated vaccine varicella zoster virus in a healthy child. Eur. J. Pediatr. 2002, 161, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.F.; Schmid, D.S.; Harpaz, R.; La Russa, P.; Jensen, N.J.; Rivailler, P.; Radford, K.; Folster, J.; Jacobsen, S.J. Herpes zoster caused by vaccine-strain varicella zoster virus in an immunocompetent recipient of zoster vaccine. Clin. Infect. Dis. 2014, 58, 1125–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fusco, D.; Krawitz, P.; La Russa, P.; Steinberg, S.; Gershon, A.; Jacobs, J. VZV meningitis following varicella vaccine. J. Clin. Virol. 2010, 48, 275–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.Y.; Hanson, D.C.; Way, S.S. Herpes zoster and meningitis due to reactivation of varicella vaccine virus in an immunocompetent child. Pediatr. Infect. Dis. J. 2011, 30, 266–268. [Google Scholar] [CrossRef]
- Chouliaras, G.; Spoulou, V.; Quinlivan, M.; Breuer, J.; Theodoridou, M. Vaccine-associated herpes zoster ophthalmicus [correction of opthalmicus] and encephalitis in an immunocompetent child. Pediatrics 2010, 125, e969–e972. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.; Broom, J. Vaccine strain varicella zoster virus transmitted within a family from a child with shingles results in varicella meningitis in an immunocompetent adult. Intern. Med. J. 2019, 49, 132–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pahwa, S.; Biron, K.; Lim, W.; Swenson, P.; Kaplan, M.H.; Sadick, N.; Pahwa, R. Continuous varicella-zoster infection associated with acyclovir resistance in a child with AIDS. JAMA 1988, 260, 2879–2882. [Google Scholar] [CrossRef]
- Snoeck, R.; Gerard, M.; Delvaux, S.C.; Andrei, G.; Balzarini, J.; Reymen, D.; Ahadi, N.; De Bruyn, J.M.; Piette, J.; Rentier, B.; et al. Meningoradiculoneuritis due to acyclovir-resistant varicella zoster virus in an acquired immune deficiency syndrome patient. J. Med. Virol. 1994, 42, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Edelman, C.K.; Pedneault, L.; Talarico, C.L.; Biron, K.K.; Balfour, H.H., Jr. Phenotypic and genotypic characterization of acyclovir-resistant varicella-zoster viruses isolated from persons with AIDS. J. Infect. Dis. 1994, 170, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Talarico, C.L.; Phelps, W.C.; Biron, K.K. Analysis of the thymidine kinase genes from acyclovir-resistant mutants of varicella-zoster virus isolated from patients with AIDS. J. Virol. 1993, 67, 1024–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piret, J.; Boivin, G. Antiviral resistance in herpes simplex virus and varicella-zoster virus infections: Diagnosis and management. Curr. Opin. Infect. Dis. 2016, 29, 654–662. [Google Scholar] [CrossRef] [PubMed]
- van der Beek, M.T.; Vermont, C.L.; Bredius, R.G.; Marijt, E.W.; van der Blij-de Brouwer, C.S.; Kroes, A.C.; Claas, E.C.; Vossen, A.C. Persistence and Antiviral Resistance of Varicella Zoster Virus in Hematological Patients. Clin. Infect. Dis. 2012, 56, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Brink, A.A.; van Gelder, M.; Wolffs, P.F.; Bruggeman, C.A.; van Loo, I.H. Compartmentalization of acyclovir-resistant varicella zoster virus: Implications for sampling in molecular diagnostics. Clin. Infect. Dis. 2011, 52, 982–987. [Google Scholar] [CrossRef] [Green Version]
- Gueudry, J.; Boutolleau, D.; Gueudin, M.; Burrel, S.; Miri, A.; Bodaghi, B.; Muraine, M. Acyclovir-resistant varicella-zoster virus keratitis in an immunocompetent patient. J. Clin. Virol. 2013, 58, 318–320. [Google Scholar] [CrossRef]
- Gilbert, C.; Smith, B.J.; Boivin, G. Resistance of herpesviruses to antiviral drugs: Clinical impacts and molecular mechanisms. Drug Resist. Updates 2002, 5, 88–114. [Google Scholar] [CrossRef]
- Andrei, G.; Topalis, D.; Fiten, P.; McGuigan, C.; Balzarini, J.; Opdenakker, G.; Snoeck, R. In vitro-selected drug-resistant varicella-zoster virus mutants in the thymidine kinase and DNA polymerase genes yield novel phenotype-genotype associations and highlight differences between antiherpesvirus drugs. J. Virol. 2012, 86, 2641–2652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Thymidine kinase and protein kinase in drug-resistant herpesviruses: Heads of a Lernaean Hydra. Drug Resist. Updates 2018, 37, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Morfin, F.; Thouvenot, D.; Tessier, T.M.; Lina, B.; Aymard, M.; Ooka, T. Phenotypic and genetic characterization of thymidine kinase from clinical strains of varicella-zoster virus resistant to acyclovir. Antimicrob. Agents Chemother. 1999, 43, 2412–2416. [Google Scholar] [CrossRef] [Green Version]
- Piret, J.; Boivin, G. Antiviral drug resistance in herpesviruses other than cytomegalovirus. Rev. Med. Virol. 2014, 24, 186–218. [Google Scholar] [CrossRef]
- Hatchette, T.; Tipples, G.A.; Peters, G.; Alsuwaidi, A.; Zhou, J.; Mailman, T.L. Foscarnet salvage therapy for acyclovir-resistant varicella zoster: Report of a novel thymidine kinase mutation and review of the literature. Pediatr. Infect. Dis. J. 2008, 27, 75–77. [Google Scholar] [CrossRef]
- Safrin, S.; Berger, T.G.; Gilson, I.; Wolfe, P.R.; Wofsy, C.B.; Mills, J.; Biron, K.K. Foscarnet therapy in five patients with AIDS and acyclovir-resistant varicella-zoster virus infection. Ann. Intern. Med. 1991, 115, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Bryan, C.J.; Prichard, M.N.; Daily, S.; Jefferson, G.; Hartline, C.; Cassady, K.A.; Hilliard, L.; Shimamura, M. Acyclovir-resistant chronic verrucous vaccine strain varicella in a patient with neuroblastoma. Pediatr. Infect. Dis. J. 2008, 27, 946–948. [Google Scholar] [CrossRef]
- Visse, B.; Dumont, B.; Huraux, J.M.; Fillet, A.M. Single amino acid change in DNA polymerase is associated with foscarnet resistance in a varicella-zoster virus strain recovered from a patient with AIDS. J. Infect. Dis. 1998, 178, S55–S57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillet, A.M.; Visse, B.; Caumes, E.; Dumont, B.; Gentilini, M.; Huraux, J.M. Foscarnet-resistant multidermatomal zoster in a patient with AIDS. Clin. Infect. Dis. 1995, 21, 1348–1349. [Google Scholar] [CrossRef]
- Visse, B.; Huraux, J.M.; Fillet, A.M. Point mutations in the varicella-zoster virus DNA polymerase gene confers resistance to foscarnet and slow growth phenotype. J. Med. Virol. 1999, 59, 84–90. [Google Scholar] [CrossRef]
- Andrei, G.; De Clercq, E.; Snoeck, R. In vitro selection of drug-resistant varicella-zoster virus (VZV) mutants (OKA strain): Differences between acyclovir and penciclovir? Antivir. Res. 2004, 61, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R.; Bacon, T.H.; Sutton, D.; Cole, M. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine (BRL 39123) in cell culture. Antimicrob. Agents Chemother. 1987, 31, 1238–1242. [Google Scholar] [CrossRef] [Green Version]
- Pelosi, E.; Mulamba, G.B.; Coen, D.M. Penciclovir and pathogenesis phenotypes of drug-resistant Herpes simplex virus mutants. Antivir. Res. 1998, 37, 17–28. [Google Scholar] [CrossRef]
- Hasegawa, T.; Kurokawa, M.; Yukawa, T.A.; Horii, M.; Shiraki, K. Inhibitory action of acyclovir (ACV) and penciclovir (PCV) on plaque formation and partial cross-resistance of ACV-resistant varicella-zoster virus to PCV. Antivir. Res. 1995, 27, 271–279. [Google Scholar] [CrossRef]
- Ida, M.; Kageyama, S.; Sato, H.; Kamiyama, T.; Yamamura, J.; Kurokawa, M.; Morohashi, M.; Shiraki, K. Emergence of resistance to acyclovir and penciclovir in varicella-zoster virus and genetic analysis of acyclovir-resistant variants. Antivir. Res. 1999, 40, 155–166. [Google Scholar] [CrossRef]
- Stryjewski, T.P.; Scott, N.L.; Barshak, M.B.; Tobin, E.H.; Mali, J.O.; Young, L.H.; Foster, C.S.; Kim, I.K.; Durand, M.L. Treatment of Refractory Acute Retinal Necrosis with Intravenous Foscarnet or Cidofovir. Ocul. Immunol. Inflamm. 2018, 26, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.L.; James, S.H. Antiviral Therapies for Herpesviruses: Current Agents and New Directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef] [Green Version]
- Levin, M.J.; Dahl, K.M.; Weinberg, A.; Giller, R.; Patel, A.; Krause, P.R. Development of resistance to acyclovir during chronic infection with the Oka vaccine strain of varicella-zoster virus, in an immunosuppressed child. J. Infect. Dis. 2003, 188, 954–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuigan, C.; Yarnold, C.J.; Jones, G.; Velazquez, S.; Barucki, H.; Brancale, A.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J. Potent and selective inhibition of varicella-zoster virus (VZV) by nucleoside analogues with an unusual bicyclic base. J. Med. Chem. 1999, 42, 4479–4484. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Barucki, H.; Carangio, A.; Blewett, S.; Andrei, G.; Snoeck, R.; De Clercq, E.; Balzarini, J.; Erichsen, J.T. Highly potent and selective inhibition of varicella-zoster virus by bicyclic furopyrimidine nucleosides bearing an aryl side chain. J. Med. Chem. 2000, 43, 4993–4997. [Google Scholar] [CrossRef]
- Andrei, G.; Sienaert, R.; McGuigan, C.; De Clercq, E.; Balzarini, J.; Snoeck, R. Susceptibilities of several clinical Varicella-zoster virus (VZV) isolates and drug-resistant VZV strains to bicyclic furano pyrimidine nucleosides. Antimicrob. Agents Chemother. 2005, 49, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzarini, J.; McGuigan, C. Chemotherapy of varicella-zoster virus by a novel class of highly specific anti-VZV bicyclic pyrimidine nucleosides. Biochim. Biophys. Acta 2002, 1587, 287–295. [Google Scholar] [CrossRef] [Green Version]
- McGuigan, C.; Pathirana, R.N.; Migliore, M.; Adak, R.; Luoni, G.; Jones, A.T.; Torrubia, D.A.; Camarasa, M.J.; Velazquez, S.; Henson, G.; et al. Preclinical development of bicyclic nucleoside analogues as potent and selective inhibitors of varicella zoster virus. J. Antimicrob. Chemother. 2007, 60, 1316–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sienaert, R.; Naesens, L.; Brancale, A.; Carangio, A.; Andrei, G.; Snoeck, R.; Van Kuilenburg, A.; De Clercq, E.; McGuigan, C.; Balzarini, J. Metabolic and pharmacological characteristics of the bicyclic nucleoside analogues (BCNAs) as highly selective inhibitors of varicella-zoster virus (VZV). Nucleosides Nucleotides Nucleic Acids 2003, 22, 995–997. [Google Scholar] [CrossRef]
- Balzarini, J.; Sienaert, R.; Liekens, S.; Van Kuilenburg, A.; Carangio, A.; Esnouf, R.; De Clercq, E.; McGuigan, C. Lack of susceptibility of bicyclic nucleoside analogs, highly potent inhibitors of varicella-zoster virus, to the catabolic action of thymidine phosphorylase and dihydropyrimidine dehydrogenase. Mol. Pharmacol. 2002, 61, 1140–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sienaert, R.; Naesens, L.; Brancale, A.; De Clercq, E.; McGuigan, C.; Balzarini, J. Specific recognition of the bicyclic pyrimidine nucleoside analogs, a new class of highly potent and selective inhibitors of varicella-zoster virus (VZV), by the VZV-encoded thymidine kinase. Mol. Pharmacol. 2002, 61, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentikis, H.S.; Matson, M.; Atiee, G.; Boehlecke, B.; Hutchins, J.T.; Patti, J.M.; Henson, G.W.; Morris, A. Pharmacokinetics and safety of FV-100, a novel oral anti-herpes zoster nucleoside analogue, administered in single and multiple doses to healthy young adult and elderly adult volunteers. Antimicrob. Agents Chemother. 2011, 55, 2847–2854. [Google Scholar] [CrossRef] [Green Version]
- Tyring, S.K.; Lee, P.; Hill, G.T., Jr.; Silverfield, J.C.; Moore, A.Y.; Matkovits, T.; Bolyai, S.J. FV-100 versus valacyclovir for the prevention of post-herpetic neuralgia and the treatment of acute herpes zoster-associated pain: A randomized-controlled trial. J. Med. Virol. 2017, 89, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Abele, G.; Eriksson, B.; Harmenberg, J.; Wahren, B. Inhibition of varicella-zoster virus-induced DNA polymerase by a new guanosine analog, 9-(4-hydroxy-2-(hydroxymethyl)butyl)guanine triphosphate. Antimicrob. Agents Chemother. 1988, 32, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Lowe, D.M.; Alderton, W.K.; Ellis, M.R.; Parmar, V.; Miller, W.H.; Roberts, G.B.; Fyfe, J.A.; Gaillard, R.; Ertl, P.; Snowden, W.; et al. Mode of action of (R)-9-(4-hydroxy-2-(hydroxymethyl)butyl)guanine against herpesviruses. Antimicrob. Agents Chemother. 1995, 39, 1802–1808. [Google Scholar] [CrossRef] [Green Version]
- Ng, T.I.; Shi, Y.; Huffaker, H.J.; Kati, W.; Liu, Y.; Chen, C.M.; Lin, Z.; Maring, C.; Kohlbrenner, W.E.; Molla, A. Selection and characterization of varicella-zoster virus variants resistant to (R)-9-(4-hydroxy-2-(hydroxymethy)butyl)guanine. Antimicrob. Agents Chemother. 2001, 45, 1629–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neyts, J.; Andrei, G.; De Clercq, E. The antiherpesvirus activity of H2G ((R)-9-(4-hydroxy-2-(hydroxymethyl)butyl)guanine) is markedly enhanced by the novel immunosuppressive agent mycophenolate mofetil. Antimicrob. Agents Chemother. 1998, 42, 3285–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Clercq, E.; Field, H.J. Antiviral prodrugs-the development of successful prodrug strategies for antiviral chemotherapy. Br. J. Pharmacol. 2006, 147, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tyring, S.K.; Plunkett, S.; Scribner, A.R.; Broker, R.E.; Herrod, J.N.; Handke, L.T.; Wise, J.M.; Martin, P.A. Valomaciclovir versus valacyclovir for the treatment of acute herpes zoster in immunocompetent adults: A randomized, double-blind, active-controlled trial. J. Med. Virol. 2012, 84, 1224–1232. [Google Scholar] [CrossRef]
- Balfour, H.H., Jr.; Vezina, H.E.; Hogquist, R.C.; Brundage, R.C.; Anderson, B.J.; Odumade, O.A. Randomized, placebo-controlled, double-blind trial of valomacicovir (VALM) for infectious mononucleosis (IM). In Proceedings of the 49th Annual Meeting of the Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, USA, 14 September 2009; p. 221. [Google Scholar]
- Hostetler, K.Y. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: Current state of the art. Antiviral. Res. 2009, 82, A84–A98. [Google Scholar] [CrossRef] [Green Version]
- Beadle, J.R.; Hartline, C.; Aldern, K.A.; Rodriguez, N.; Harden, E.; Kern, E.R.; Hostetler, K.Y. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob. Agents Chemother. 2002, 46, 2381–2386. [Google Scholar] [CrossRef] [Green Version]
- Marty, F.M.; Winston, D.J.; Chemaly, R.F.; Mullane, K.M.; Shore, T.B.; Papanicolaou, G.A.; Chittick, G.; Brundage, T.M.; Wilson, C.; Morrison, M.E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transplant. 2019, 25, 369–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Neofytos, D.; Kim, S.J.; Cheteyan, L.; Huang, Y.T.; Papadopoulos, E.B.; Jakubowski, A.A.; Papanicolaou, G.A. Efficacy of brincidofovir as prophylaxis against HSV and VZV in hematopoietic cell transplant recipients. Transpl. Infect. Dis. 2018, 20, 2977. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K.M.; Nuss, C.; Ridgeway, J.; Prichard, M.N.; Hartline, C.B.; Theusch, J.; Marin, M.H.; Larson, R.A. Brincidofovir treatment of acyclovir-resistant disseminated varicella zoster virus infection in an immunocompromised host. Transpl. Infect. Dis. 2016, 18, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Katsamakas, S.; Zografos, A.L.; Sarli, V. Advances of Phenoxazines: Synthesis, Reactivity and Their Medicinal Applications. Curr. Med. Chem. 2016, 23, 2972–2999. [Google Scholar] [CrossRef] [PubMed]
- Kozlovskaya, L.I.; Andrei, G.; Orlov, A.A.; Khvatov, E.V.; Koruchekov, A.A.; Belyaev, E.S.; Nikolaev, E.N.; Korshun, V.A.; Snoeck, R.; Osolodkin, D.I.; et al. Antiviral activity spectrum of phenoxazine nucleoside derivatives. Antivir. Res. 2019, 163, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Hayashi, T.; Tomoda, A. Phenoxazine derivatives inactivate human cytomegalovirus, herpes simplex virus-1, and herpes simplex virus-2 in vitro. J. Pharmacol. Sci. 2008, 106, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Plebanek, E.; Lescrinier, E.; Andrei, G.; Snoeck, R.; Herdewijn, P.; De Jonghe, S. Emimycin and its nucleoside derivatives: Synthesis and antiviral activity. Eur. J. Med. Chem. 2018, 144, 93–103. [Google Scholar] [CrossRef]
- Sari, O.; Roy, V.; Balzarini, J.; Snoeck, R.; Andrei, G.; Agrofoglio, L.A. Synthesis and antiviral evaluation of C5-substituted-(1,3-diyne)-2′-deoxyuridines. Eur. J. Med. Chem. 2012, 53, 220–228. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Q.; Yang, M.; Schneller, S.W. C-3 halo and 3-methyl substituted 5′-nor-3-deazaaristeromycins: Synthesis and antiviral properties. Bioorg. Med. Chem. 2013, 21, 359–364. [Google Scholar] [CrossRef]
- Baszczynski, O.; Kaiser, M.M.; Cesnek, M.; Brehova, P.; Jansa, P.; Prochazkova, E.; Dracinsky, M.; Snoeck, R.; Andrei, G.; Janeba, Z. Xanthine-based acyclic nucleoside phosphonates with potent antiviral activity against varicella-zoster virus and human cytomegalovirus. Antivir. Chem. Chemother. 2018, 26, 3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Srivastava, P.; Liu, C.; Snoeck, R.; Andrei, G.; De Jonghe, S.; Herdewijn, P. Synthesis and antiviral evaluation of cyclopentyl nucleoside phosphonates. Eur. J. Med. Chem. 2018, 150, 616–625. [Google Scholar] [CrossRef]
- Bessieres, M.; Hervin, V.; Roy, V.; Chartier, A.; Snoeck, R.; Andrei, G.; Lohier, J.F.; Agrofoglio, L.A. Highly convergent synthesis and antiviral activity of (E)-but-2-enyl nucleoside phosphonoamidates. Eur. J. Med. Chem. 2018, 146, 678–686. [Google Scholar] [CrossRef]
- Topalis, D.; Pradere, U.; Roy, V.; Caillat, C.; Azzouzi, A.; Broggi, J.; Snoeck, R.; Andrei, G.; Lin, J.; Eriksson, S.; et al. Novel antiviral C5-substituted pyrimidine acyclic nucleoside phosphonates selected as human thymidylate kinase substrates. J. Med. Chem. 2011, 54, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Pertusati, F.; Serafini, S.; Albadry, N.; Snoeck, R.; Andrei, G. Phosphonoamidate prodrugs of C5-substituted pyrimidine acyclic nucleosides for antiviral therapy. Antivir. Res. 2017, 143, 262–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krecmerova, M.; Dracinsky, M.; Snoeck, R.; Balzarini, J.; Pomeisl, K.; Andrei, G. New prodrugs of two pyrimidine acyclic nucleoside phosphonates: Synthesis and antiviral activity. Bioorg. Med. Chem. 2017, 25, 4637–4648. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Groaz, E.; Andrei, G.; Snoeck, R.; Kalkeri, R.; Ptak, R.G.; Hartman, T.; Buckheit, R.W., Jr.; Schols, D.; De Jonghe, S.; et al. Expanding the Antiviral Spectrum of 3-Fluoro-2-(phosphonomethoxy)propyl Acyclic Nucleoside Phosphonates: Diamyl Aspartate Amidate Prodrugs. J. Med. Chem. 2017, 60, 6220–6238. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Groaz, E.; De Jonghe, S.; Snoeck, R.; Andrei, G.; Herdewijn, P. Amidate Prodrugs of Cyclic 9-(S)-[3-Hydroxy-2-(phosphonomethoxy)propyl]adenine with Potent Anti-Herpesvirus Activity. ACS Med. Chem. Lett. 2018, 9, 381–385. [Google Scholar] [CrossRef]
- Inoue, N.; Matsushita, M.; Fukui, Y.; Yamada, S.; Tsuda, M.; Higashi, C.; Kaneko, K.; Hasegawa, H.; Yamaguchi, T. Identification of a varicella-zoster virus replication inhibitor that blocks capsid assembly by interacting with the floor domain of the major capsid protein. J. Virol. 2012, 86, 12198–12207. [Google Scholar] [CrossRef] [Green Version]
- Yasui, R.; Yoshida, C.; Yamaguchi, T.; Inoue, N. Characterization of an anti-varicella-zoster virus compound that targets the portal protein encoded by ORF54. Microbiol. Immunol. 2017, 61, 398–402. [Google Scholar] [CrossRef] [Green Version]
- Majima, R.; Shindoh, K.; Yamaguchi, T.; Inoue, N. Characterization of a thienylcarboxamide derivative that inhibits the transactivation functions of cytomegalovirus IE2 and varicella zoster virus IE62. Antivir. Res. 2017, 140, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Musella, S.; di Sarno, V.; Ciaglia, T.; Sala, M.; Spensiero, A.; Scala, M.C.; Ostacolo, C.; Andrei, G.; Balzarini, J.; Snoeck, R.; et al. Identification of an indol-based derivative as potent and selective varicella zoster virus (VZV) inhibitor. Eur. J. Med. Chem. 2016, 124, 773–781. [Google Scholar] [CrossRef]
- Kim, J.E.; Song, Y.J. Anti-varicella-zoster virus activity of cephalotaxine esters in vitro. J. Microbiol. 2019, 57, 74–79. [Google Scholar] [CrossRef]
- Kaoukabi, H.; Kabri, Y.; Curti, C.; Taourirte, M.; Ubis, R.J.C.; Snoeck, R.; Andrei, G.; Vanelle, P.; Lazrek, H.B. Dihydropyrimidinone/1,2,3-triazole hybrid molecules: Synthesis and anti-varicella-zoster virus (VZV) evaluation. Eur. J. Med. Chem. 2018, 155, 772–781. [Google Scholar] [CrossRef]
- Gupta, A.; Dixit, B.; Stamoulas, K.; Akshikar, R. Atypical bilateral acute retinal necrosis in a coronavirus disease 2019 positive immunosuppressed patient. Eur. J. Ophthalmol. 2020, 4941. [Google Scholar] [CrossRef]
- Tartari, F.; Spadotto, A.; Zengarini, C.; Zanoni, R.; Guglielmo, A.; Adorno, A.; Valzania, C.; Pileri, A. Herpes zoster in COVID-19-positive patients. Int. J. Dermatol. 2020, 59, 1028–1029. [Google Scholar] [CrossRef] [PubMed]
- Shors, A.R. Herpes zoster and severe acute herpetic neuralgia as a complication of COVID-19 infection. JAAD Case Rep. 2020, 6, 656–657. [Google Scholar] [CrossRef]
- Elsaie, M.L.; Youssef, E.A.; Nada, H.A. Herpes zoster might be an indicator for latent COVID 19 infection. Dermatol. Ther. 2020, 33, 3666. [Google Scholar] [CrossRef]
- Ferreira, A.; Romao, T.T.; Macedo, Y.S.; Pupe, C.; Nascimento, O.J.M.; Fellow of the American Academy of Neurology (FAAN). COVID-19 and herpes zoster co-infection presenting with trigeminal neuropathy. Eur. J. Neurol. 2020, 27, 1748–1750. [Google Scholar] [CrossRef] [PubMed]
- Saati, A.; Husayni, A.F.; Malibari, A.A.; Bogari, A.A.; Alharbi, M. Herpes Zoster Co-Infection in an Immunocompetent Patient with COVID-19. Cureus 2020, 12, 8998. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Varicella Vaccine | Herpes Zoster Vaccine | ||
|---|---|---|---|
| Name | Varivax® & Varilrix® | Zostavax® | Shingrix® |
| Year of FDA licensure | 1995 | 2006 | 2018 |
| Manufacturer | Merck (Varivax®) GSK (Varilrix®) | Merck | GSK |
| Type | Life-attenuated viral vaccine | Life-attenuated viral vaccine | Inactivated; Recombinant subunit |
| Vaccine components | Oka strain (1350 PFU) | Oka strain (19,400 PFU) | VZV glycoprotein E (gE) |
| Number of doses administered |
| 1 dose | 2 doses (2–6 months apart) |
| Storage | Freezer | Freezer | Refrigerator |
| Diluent | Sterile water | Sterile water | Adjuvant |
| Dose form | 0.5 mL vial for intramuscular injection | 0.65 mL vial for subcutaneous injection | 0.5 mL vial for intramuscular injection |
| Recommended age |
|
|
|
| Efficacy | About 98% protection in children and about 75% protection in teenagers and adults |
|
|
| Immunity duration | Unknown; however, long-term efficacy studies have demonstrated continued protection up to 10 years after vaccination | Age-dependent | Age-dependent |
| Contraindications |
|
|
|
| Possible side effects | Most commonly:
| Most commonly:
| Most commonly:
|
| Drug | Prodrug | Mode of Action/Approval/Indication | Natural Analogue |
|---|---|---|---|
| I. Nucleoside analogues | |||
 |  |
|  |
 |  |
|  |
 | Orally bioavailable |
|  |
| II. Nucleotide analogue | |||
 |  |
|  |
| III. Pyrophosphate analogue | |||
 |
| ||
| IV. Non-nucleoside analogue | |||
 | Orally bioavailable |
| |
| Compound | EC50 TK+ VZV | EC50 TK− VZV | CC50 | Activity against Other Human Herpesviruses | |
|---|---|---|---|---|---|
| Nucleoside/nucleotide analogues | |||||
 | 0.06 µM | 10 µM | >100 µM | No activity against HCMV | |
 | 0.99 µM | 6.91 µM | >412 µM | Activity against HSV-1, but lower activity against TK− HSV-1 and against HSV-2. No anti-HCMV activity | |
 | ~1 µM | >20 µM | 55 µM | Decreased activity against HSV-1 TK− and HSV-2.No anti-HCMV activity | |
 | 0.11 µM | No data available | >300 µM | Activity against HCMV, no data on other herpesviruses | |
 | 2.62 µM | 4.58 µM | 111 µM | Activity against HSV-1, HSV-2 and HCMV | |
 | 5.35 µM | 8.83 µM | >289 µM | Activity against HSV-1, HSV-2 and HCMV | |
| Nuceoside/nucleotide prodrugs | |||||
 | 0.39 µM | 0.33 µM | ≥83 µM | Activity against HSV-1, HSV-2 and HCMV | |
| Prodrugs of C5-substituted pyrimidine acyclic nucleosides Bis(POM) derivative of (E)-TbutP (compound 16) Bis(POM) derivative of (E)-5Br-UbutP (compound 18) | |||||
 | 0.41 µM | 0.19 µM | 35 µM | Activity against HSV-1 & HSV-2 and no anti-HCMV activity | |
| R = Br (E)- compound 18 | 20 µM | 15 µM | 35 µM | Weak activity against HSV-1, HSV-2, no anti-HCMV activity | |
 | 0.5 µM | 0.09 µM | 40 µM | Activity against HSV-1, HSV-2 and HCMV | |
 | 0.47 µM | 0.20 µM | >100 µM | Activity against HSV-1, HSV-2 and HCMV | |
| Prodrugs of pyrimidine acyclic nucleoside analogues | |||||
 | 2.07 µM | 4.72 µM | 90.3 µM | Activity against HSV-1 & HSV-2 but not against HCMV | |
 | 0.32 µM | 0.29 µM | 15.1 µM | Activity against HSV-1 & HSV-2 and marginal activity against HCMV | |
 | 0.56 µM | 1.69 µM | 25.9 µM | Activity against HSV-1 & HSV-2 and low activity against HCMV | |
 | 100 µM | 40 µM | >200 µM | Marginal activity against HSV-1, HSV-2 & HCMV | |
 | 0.32 µM | 0.79 µM | 79.2 µM | Activity against HSV-1, HSV-2 and HCMV | |
 | |||||
| 22a | 0.05 µM | 0.057 µM | 1.96 µM | No selectivity against HCMV | |
| 23a | 3.15 µM | 1.32 µM | 74.7 µM | Activity and selectivity against HCMV | |
| 25a | 0.59 µM | 0.079 µM | 27.8 µM | Activity but low selectivity against HCMV | |
| 25b | 0.89 µM | 0.11 µM | 10.2 µM | ||
 | 0.00045 µM | 0.00054 µM | 12 µM | Activity against HSV-1, HSV-2, and HCMV | |
| Non-nucleoside analogues | |||||
 | 0.75 µM | 4.6 µM | 152.5 µM | Weak activity against HSV-1, no anti-HCMV activity | |
 | 16.9 µM | 18.3 µM | >100 µM | No data available on other viruses | |
 | 19.3 µM | No data available | 537 µM | Activity against HCMV | |
 | 2.6 µM | 2.0 µM | >100 µM | No activity against HSV-1, HSV-2, HCMV | |
| Cephalotaxine esters | |||||
 |  | 16.15 ng/mL (HT) 9.96 ng/mL (HTT) | No data | 45.82 ng/mL (HT) 74.71 ng/mL (HTT) | No data available on other viruses—Low selectivity for VZV |
| HT | HTT | ||||
| Hybrid molecules | |||||
| Dihydropyrimidinone/1,2,3-triazole hybrid molecules | |||||
 |  | 3.62 µM (8a) 11.33 µM (7d) | 7.85 µM (8a) 10.79 µM (7d) | >100 µM (8a) >100 µM (7d) | No anti-HCMV activity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrei, G.; Snoeck, R. Advances and Perspectives in the Management of Varicella-Zoster Virus Infections. Molecules 2021, 26, 1132. https://doi.org/10.3390/molecules26041132
Andrei G, Snoeck R. Advances and Perspectives in the Management of Varicella-Zoster Virus Infections. Molecules. 2021; 26(4):1132. https://doi.org/10.3390/molecules26041132
Chicago/Turabian StyleAndrei, Graciela, and Robert Snoeck. 2021. "Advances and Perspectives in the Management of Varicella-Zoster Virus Infections" Molecules 26, no. 4: 1132. https://doi.org/10.3390/molecules26041132
APA StyleAndrei, G., & Snoeck, R. (2021). Advances and Perspectives in the Management of Varicella-Zoster Virus Infections. Molecules, 26(4), 1132. https://doi.org/10.3390/molecules26041132