
Structural and Thermodynamic Analysis of the Resistance Development to Pimodivir (VX-787), the Clinical Inhibitor of Cap Binding to PB2 Subunit of Influenza A Polymerase †


 ,
,  , and
, and
Abstract
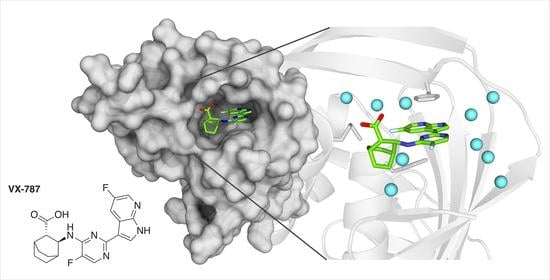
:
1. Introduction
2. Results and Discussion
2.1. The Mutated Forms M431I, F404Y and H357N of PB2 Impair Its Interaction with Pimodivir
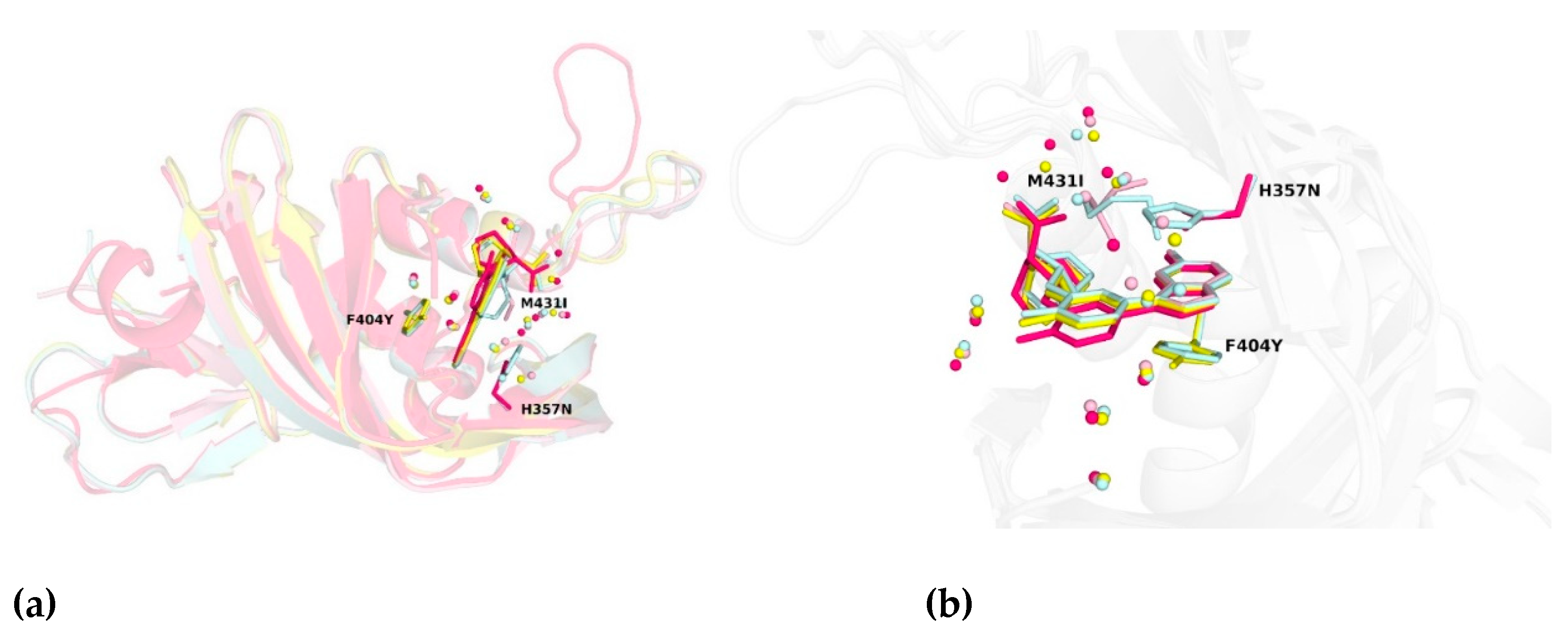
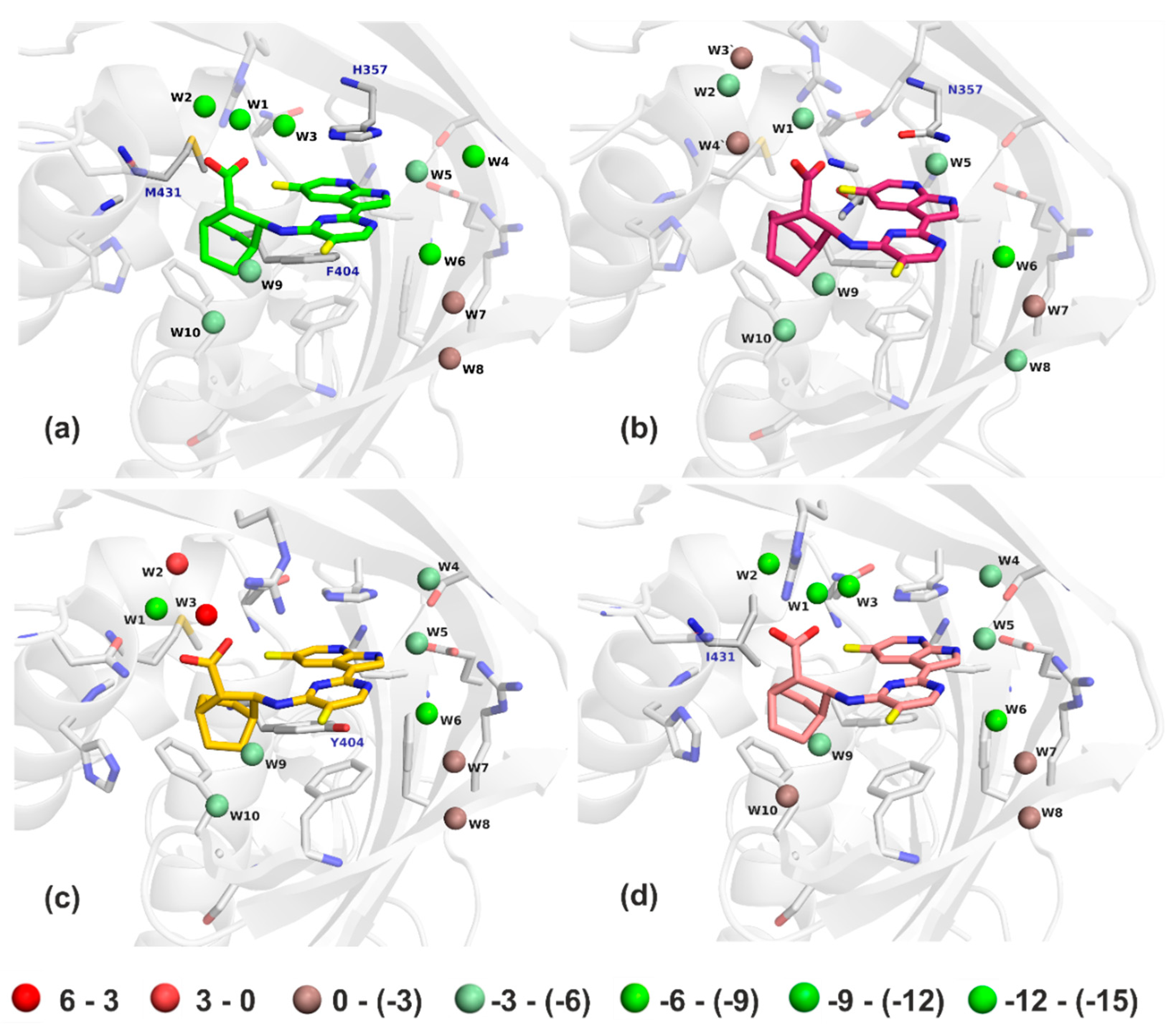
2.2. Crystal Structures Illustrate Influence of Mutations in PB2 on Pimodivir Binding and Help Elucidate the Mechanism of Pimodivir Resistance
2.3. Quantum Mechanical Analysis and Subsequent Modelling of PB2 Variants
3. Materials and Methods
3.1. Cloning, Expression, and Purification of Recombinant Proteins
3.2. Isothermal Titration Calorimetry
3.3. Protein Crystallization
3.4. Data Collection and Structure Determination
3.5. Structural Analysis of Pimodivir Binding to PB2 Variants
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taubenberger, J.K.; Kash, J.C. Influenza Virus Evolution, Host Adaptation, and Pandemic Formation. Cell Host Microbe 2010, 7, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertz, D.; Kim, T.H.; Johnstone, J.; Lam, P.-P.; Science, M.; Kuster, S.P.; Fadel, S.; Tran, D.; Fernandez, E.; Bhatnagar, N.; et al. Populations at risk for severe or complicated influenza illness: Systematic review and meta-analysis. BMJ 2013, 347, f5061. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Xie, Y.; Shi, L.; Xu, W. Viral RNA polymerase: A promising antiviral target for influenza A virus. Curr. Med. Chem. 2013, 20, 3923–3934. [Google Scholar] [CrossRef]
- Peng, Q.; Liu, Y.; Peng, R.; Wang, M.; Yang, W.; Song, H.; Chen, Y.; Liu, S.; Han, M.; Zhang, X.; et al. Structural insight into RNA synthesis by influenza D polymerase. Nat. Microbiol. 2019, 4, 1750–1759. [Google Scholar] [CrossRef]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nat. Cell Biol. 2014, 516, 355–360. [Google Scholar] [CrossRef]
- Dias, A.T.; Bouvier, D.; Crépin, T.; McCarthy, A.A.; Hart, D.J.; Baudin, F.; Cusack, S.; Ruigrok, R.W.H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nat. Cell Biol. 2009, 458, 914–918. [Google Scholar] [CrossRef]
- Reich, S.; Guilligay, D.; Pflug, A.; Malet, H.; Berger, I.; Crépin, T.; Hart, D.J.; Lunardi, T.; Nanao, M.; Ruigrok, R.W.H.; et al. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nat. Cell Biol. 2014, 516, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Hayden, F.G.; Shindo, N. Influenza virus polymerase inhibitors in clinical development. Curr. Opin. Infect. Dis. 2019, 32, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.P.; Ledeboer, M.W.; Davies, I.; Byrn, R.A.; Jones, S.M.; Perola, E.; Tsai, A.; Jacobs, M.; Nti-Addae, K.; Bandarage, U.K.; et al. Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2. J. Med. Chem. 2014, 57, 6668–6678. [Google Scholar] [CrossRef] [PubMed]
- Byrn, R.A.; Jones, S.M.; Bennett, H.B.; Bral, C.; Clark, M.P.; Jacobs, M.D.; Kwong, A.D.; Ledeboer, M.W.; Leeman, J.R.; McNeil, C.F.; et al. Preclinical Activity of VX-787, a First-in-Class, Orally Bioavailable Inhibitor of the Influenza Virus Polymerase PB2 Subunit. Antimicrob. Agents Chemother. 2015, 59, 1569–1582. [Google Scholar] [CrossRef] [Green Version]
- Finberg, R.W.; Lanno, R.; Anderson, D.; Fleischhackl, R.; Van Duijnhoven, W.; Kauffman, R.S.; Kosoglou, T.; Vingerhoets, J.; Leopold, L. Phase 2b Study of Pimodivir (JNJ-63623872) as Monotherapy or in Combination with Oseltamivir for Treatment of Acute Uncomplicated Seasonal Influenza A: TOPAZ Trial. J. Infect. Dis. 2018, 219, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubareva, L.V.; Bethell, R.; Hart, G.J.; Murti, K.G.; Penn, C.R.; Webster, R.G. Characterization of mutants of influenza A virus selected with the neuraminidase inhibitor 4-guanidino-Neu5Ac2en. J. Virol. Mar. 1996, 70, 1818–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubareva, L.; Mishin, V.; Patel, M.; Chesnokov, A.; Cruz, J.D.L.; Nguyen, H.; Lollis, L.; Hodges, E.; Jang, Y.; Barnes, J.; et al. Seasonal and other influenza viruses with reduced susceptibility to Baloxavir and Pimodivir. In Proceedings of the OPTIONS X for the Control of Influenza, Suntec City, Singapore, 28 August–1 September 2019. No. 10750. [Google Scholar]
- Zhu, W.; Zhu, Y.; Qin, K.; Yu, Z.; Gao, R.; Yu, H.; Zhou, J.; Shu, Y. Mutations in Polymerase Genes Enhanced the Virulence of 2009 Pandemic H1N1 Influenza Virus in Mice. PLoS ONE 2012, 7, e33383. [Google Scholar] [CrossRef] [PubMed]
- Fechter, P.; Mingay, L.; Sharps, J.; Chambers, A.; Fodor, E.; Brownlee, G.G. Two Aromatic Residues in the PB2 Subunit of Influenza a RNA Polymerase Are Crucial for Cap Binding. J. Biol. Chem. 2003, 278, 20381–20388. [Google Scholar] [CrossRef] [Green Version]
- Trevejo, J.M.; Asmal, M.; Vingerhoets, J.; Polo, R.; Robertson, S.; Jiang, Y.; Kieffer, T.L.; Leopold, L. Pimodivir treatment in adult volunteers experimentally inoculated with live influenza virus: A Phase IIa, randomized, double-blind, placebo-controlled study. Antivir Ther. 2018, 23, 335–344. [Google Scholar] [CrossRef]
- Lee, S.; Jacobson, I.; Xiao, H.; Sanchez, E.; Feese, M.; Lin, B.; Adolphson, J.; Uher, L. Development of a new class of broad spectrum influenza PB2 inhibitors. In Proceedings of the OPTIONS X for the Control of Influenza, Suntec City, Singapore, 28 August–1 September 2019. No. 11025. [Google Scholar]
- Darby, J.F.; Hopkins, A.P.; Shimizu, S.; Roberts, S.M.; Brannigan, J.A.; Turkenburg, J.P.; Thomas, G.H.; Hubbard, R.E.; Fischer, M. Water Networks Can Determine the Affinity of Ligand Binding to Proteins. J. Am. Chem. Soc. 2019, 141, 15818–15826. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Kang, K.; Sastry, M.; Sherman, W.; Sankaran, B.; Zwart, P.H.; Whitesides, G.M. Water-Restructuring Mutations Can Reverse the Thermodynamic Signature of Ligand Binding to Human Carbonic Anhydrase. Angew. Chem. Int. Ed. 2017, 56, 3833–3837. [Google Scholar] [CrossRef] [Green Version]
- Pokorná, J.; Pachl, P.; Karlukova, E.; Hejdánek, J.; Řezáčová, P.; Machara, A.; Hudlický, J.; Konvalinka, J.; Kožíšek, M. Kinetic, Thermodynamic, and Structural Analysis of Drug Resistance Mutations in Neuraminidase from the 2009 Pandemic Influenza Virus. Viruses 2018, 10, 339. [Google Scholar] [CrossRef] [Green Version]
- Krimmer, S.G.; Klebe, G. Thermodynamics of protein–ligand interactions as a reference for computational analysis: How to assess accuracy, reliability and relevance of experimental data. J. Comput. Mol. Des. 2015, 29, 867–883. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [Green Version]
- A Vagin, A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of theCCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Delano, W.L. The PyMOL Molecular Graphics System. Available online: http://www.pymol.org (accessed on 10 March 2020).
- Case, D.; Babin, V.; Berryman, J.T.; Betz, R.M. Amber 2014. 2014. Available online: http://ambermd.org (accessed on 10 June 2020).
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2010, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Fanfrlík, J.; Bronowska, A.K.; ŘezáčJ, P.; Konvalinka, J.; Hobza, P. A Reliable Docking/Scoring Scheme Based on the Semiempirical Quantum Mechanical PM6-DH2 Method Accurately Covering Dispersion and H-Bonding: HIV-1 Protease with 22 Ligands. J. Phys. Chem. B 2010, 114, 12666–12678. [Google Scholar] [CrossRef]
- Lepšík, M.; Řezáč, J.; Kolář, M.H.; Pecina, A.; Hobza, P.; Fanfrlík, J. The Semiempirical Quantum Mechanical Scoring Function for In Silico Drug Design. ChemPlusChem 2013, 78, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Pecina, A.; Eyrilmez, S.M.; Köprülüoğlu, C.; Miriyala, V.M.; Lepšík, M.; Fanfrlík, J.; Řezáč, J.; Hobza, P. SQM/COSMO Scoring Function: Reliable Quantum-Mechanical Tool for Sampling and Ranking in Structure-Based Drug Design. ChemPlusChem 2020, 85, 2362–2371. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Řezáč, J.; Hobza, P. Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods IV: Extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 2004, 10, 155–164. [Google Scholar] [CrossRef]
- Řezáč, J. Cuby: An integrative framework for computational chemistry. J. Comput. Chem. 2016, 37, 1230–1237. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J. Cuby—Ruby Framework for Computational Chemistry, Version 4. Available online: http://cuby4.molecular.cz (accessed on 15 November 2020).
- Kříž, K.; Řezáč, J. Reparametrization of the COSMO Solvent Model for Semiempirical Methods PM6 and PM7. J. Chem. Inf. Model. 2018, 59, 229–235. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stoichiometry | ΔG | ΔH | -T.ΔS | Kd | Fold | |
|---|---|---|---|---|---|---|
| PB2 Mutation a | Pimodivir/PB2 | kcal.mol−1 | kcal.mol−1 | kcal.mol−1 | nM | Kd b |
| (wild-type) | 0.97 ± 0.04 | −11.8 ± 0.1 | −11.1 ± 0.3 | −0.8 ± 0.4 | 2.2 ± 0.5 | 1 |
| F404Y | 1.04 ± 0.09 | −8.5 ± 0.1 | −6.3 ± 0.1 | −2.2 ± 0.2 | 610 ± 100 | 280 |
| M431I | 1.02 ± 0.03 | −10.7 ± 0.1 | −10.6 ± 0.2 | −0.2 ± 0.3 | 14 ± 1 | 7 |
| H357N | 1.11 ± 0.04 | −8.9 ± 0.1 | −10.6 ± 0.2 | 1.7 ± 0.3 | 290 ± 20 | 130 |
| ∆Eint | ∆∆Gsolv | ∆G’conf(L) | ∆G’conf(P) | ∆G’ | ∆G’ without ∆G’conf(P) | |
|---|---|---|---|---|---|---|
| kcal.mol−1 | kcal.mol−1 | kcal.mol−1 | kcal.mol−1 | kcal.mol−1 | kcal.mol−1 | |
| pimodivir/PB2-WT | −436.3 | 350.3 | 3.4 | 3.9 | −78.7 | −82.6 |
| pimodivir/PB2-H357N | −445.4 | 369.8 | 4.5 | 0.0 | −71.1 | −71.1 |
| pimodivir/PB2-F404Y | −394.9 | 324.4 | 5.5 | 8.3 | −56.7 | −65.0 |
| pimodivir/PB2-M431I | −444.2 | 365.4 | 3.5 | 2.2 | −73.1 | −75.3 |
| Water Molecule | Pimodivir/PB2-WT | Pimodivir/PB2-H357N | Pimodivir/PB2-F404Y | Pimodivir/PB2-M431I |
|---|---|---|---|---|
| W1 | −12.1 | −4.5 | −8.0 | −15.4 |
| W2 | −8.3 | 0.0 | 2.7 | −7.1 |
| W3 | −7.6 | −3.2 | 6.1 | −8.1 |
| W4 | −6.1 | −2.2 | −3.9 | −5.0 |
| W5 | −4.4 | −5.4 | −5.3 | −4.3 |
| W6 | −7.7 | −6.5 | −8.6 | −6.7 |
| W7 | −0.9 | −0.7 | −1.5 | −0.9 |
| W8 | −2.6 | −4.9 | −2.4 | −2.3 |
| W9 | −3.9 | −3.0 | −3.3 | −3.9 |
| W10 | −3.8 | −3.8 | −3.3 | −1.9 |
| PB2 Variant | PB2-WT | PB2-F404Y | PB2-M431I | PB2-H357N |
|---|---|---|---|---|
| PDB Code | 7AS0 | 7AS1 | 7AS2 | 7AS3 |
| Data Collection Statistics | ||||
| Wavelength (Å) | 1.5418 | 1.5418 | 1.5418 | 1.5418 |
| Space group | P1 | P1 | P1 | P3121 |
| Cell parameters (Å. o) | 29.30, 36.95, 38.34, 71.1, 75.6, 76.3 | 29.50, 37.25, 38.39, 71.9, 69.8, 75.2 | 29.19, 37.12, 38.45, 71.9, 75.4, 75.9 | 64.81, 64.8, 75.63, 90.0, 90.0, 120.0 |
| Resolution range (Å) | 50.00–1.55 (1.59–1.55) | 50.00–1.50 (1.54–1.50) | 50.00–1.75 (1.80–1.75) | 50.00–1.55 (1.59–1.55) |
| Number of unique reflections | 20014 (1471) | 22637 (1558) | 12192 (290) | 26694 (1861) |
| Multiplicity | 2.6 (1.8) | 2.5 (1.3) | 3.0 (1.4) | 5.9 (3.9) |
| Completeness (%) | 94.7 (95.3) | 98.0 (91.2) | 82.6 (26.6) | 98.2 (94.1) |
| Rmerge a | 5.4 (13.6) | 3.5 (9.3) | 3.9 (24.9) | 4.7 (122) |
| CC(1/2) (%) | 99.7 (96.8) | 99.8 (97.6) | 99.8 (76.1) | 99.9 (46.3) |
| Average I/σ(I) | 12.02 (4.27) | 18.27 (4.05) | 19.48 (2.61) | 17.10 (0.99) |
| Wilson B (Å2) | 17.92 | 21.00 | 24.20 | 30.60 |
| Refinement Statistics | ||||
| Resolution range (Å) | 35.64–1.55 (1.59–1.55) | 34.93–1.50 (1.54–1.50) | 24.34–1.750 (1.79–1.75) | 32.42–1.65 (1.69–1.65) |
| No. of reflection in working set | 19032 (996) | 21508 (1479) | 11584 (274) | 21252 (1510) |
| No. of reflection in the test set | 1394 (72) | 1129 (77) | 610 (14) | 1119 (79) |
| Rwork value (%) b | 0.183 (0.261) | 0.150 (0.179) | 0.160 (0.236) | 0.163 (0.261) |
| Rfree value (%) c | 0.220 (0.292) | 0.185 (0.230) | 0.162 (0.286) | 0.171 (0.286) |
| RMSD bond length (Å) | 0.02 | 0.01 | 0.01 | 0.01 |
| RMSD angle (o) | 1.8 | 1.9 | 1.6 | 1.5 |
| Mean ADP value (Å2) | 13.78 | 16.85 | 17.29 | 25.86 |
| Ramachandran Plot Statistics d | ||||
| Residues in favored regions (%) | 98.12 | 96.88 | 96.25 | 96.45 |
| Residues in allowed regions (%) | 1.88 | 3.12 | 3.13 | 3.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregor, J.; Radilová, K.; Brynda, J.; Fanfrlík, J.; Konvalinka, J.; Kožíšek, M. Structural and Thermodynamic Analysis of the Resistance Development to Pimodivir (VX-787), the Clinical Inhibitor of Cap Binding to PB2 Subunit of Influenza A Polymerase. Molecules 2021, 26, 1007. https://doi.org/10.3390/molecules26041007
Gregor J, Radilová K, Brynda J, Fanfrlík J, Konvalinka J, Kožíšek M. Structural and Thermodynamic Analysis of the Resistance Development to Pimodivir (VX-787), the Clinical Inhibitor of Cap Binding to PB2 Subunit of Influenza A Polymerase. Molecules. 2021; 26(4):1007. https://doi.org/10.3390/molecules26041007
Chicago/Turabian StyleGregor, Jiří, Kateřina Radilová, Jiří Brynda, Jindřich Fanfrlík, Jan Konvalinka, and Milan Kožíšek. 2021. "Structural and Thermodynamic Analysis of the Resistance Development to Pimodivir (VX-787), the Clinical Inhibitor of Cap Binding to PB2 Subunit of Influenza A Polymerase" Molecules 26, no. 4: 1007. https://doi.org/10.3390/molecules26041007
APA StyleGregor, J., Radilová, K., Brynda, J., Fanfrlík, J., Konvalinka, J., & Kožíšek, M. (2021). Structural and Thermodynamic Analysis of the Resistance Development to Pimodivir (VX-787), the Clinical Inhibitor of Cap Binding to PB2 Subunit of Influenza A Polymerase. Molecules, 26(4), 1007. https://doi.org/10.3390/molecules26041007