
New Bifunctional Bis(azairidacycle) with Axial Chirality via Double Cyclometalation of 2,2′-Bis(aminomethyl)-1,1′-binaphthyl



Abstract
:1. Introduction
2. Results and Discussion
2.1. A Short-Cut Synthesis of 2,2′-Bis(aminomethyl)-1,1′-Binaphthyl
2.2. Synthesis of Bis(azairidacycle) via Double Cyclometalation
2.3. Catalytic Application to the Asymmetric Transfer Hydrogenation of Acetophenone in 2-Propanol
3. Materials and Methods
3.1. General Information
3.2. Synthesis of 2,2′-Bis(aminomethyl)-1,1′-Binaphthyl
3.2.1. Synthesis of (S)-1,1′-Binaphthalene-2,2′-Bis(trifluoromethanesulfonate) (2)
3.2.2. Catalytic Dicyanation of 2
3.2.3. Reduction of 3
3.3. Double Cyclometalation of 1
3.4. Elimination of HCl from 5 by Treatment of KOC(CH3)3
3.5. H NMR Observation of the Generation of Hydrido(Amine)Iridium Species from 6
3.6. Catalytic Asymmetric Transfer Hydrogenation of Acetophenone In 2-Propanol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ikariya, T.; Blacker, A.J. Asymmetric transfer hydrogenation of ketones with bifunctional transition metal-based molecular catalysts. Acc. Chem. Res. 2007, 40, 1300–1308. [Google Scholar] [CrossRef]
- Ikariya, T. Chemistry of Concerto Molecular Catalysis Based on the Metal/MH Bifunctionality. Bull. Chem. Soc. Jpn. 2011, 84, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Han, Z.; Ding, K. The N-H Functional Group in Organometallic Catalysis. Angew. Chem. Int. Ed. 2013, 52, 4744–4788. [Google Scholar] [CrossRef]
- Khusnutdinova, J.R.; Milstein, D. Metal-Ligand Cooperation. Angew. Chem. Int. Ed. 2015, 54, 12236–12273. [Google Scholar] [CrossRef]
- Dub, P.A.; Gordon, J.C. The role of the metal-bound N–H functionality in Noyori-type molecular catalysts. Nat. Rev. Chem. 2018, 2, 396–408. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric Transfer Hydrogenation of Aromatic Ketones Catalyzed by Chiral Ruthenium(II) Complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. The catalyst precursor, catalyst and intermediate in the RuII-promoted asymmetric hydrogen transfer between alcohols and ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 285–288. [Google Scholar] [CrossRef]
- Kawano, T.; Watari, R.; Kayaki, Y.; Ikariya, T. Catalytic Hydrogenation of Carboxamides with a Bifunctional Cp*Ru Catalyst Bearing an Imidazol-2-ylidene with a Protic Aminoethyl Side Chain. Synthesis 2019, 51, 2542–2547. [Google Scholar] [CrossRef]
- Matsunami, A.; Kuwata, S.; Kayaki, Y. Hydrogen Evolution from Formic Acid and Hydrodefluorination of Fluoroarenes by Bifunctional Iridium Catalysts—Beyond the Transfer Hydrogenation. J. Synth. Org. Chem. Jpn. 2018, 76, 315–324. [Google Scholar] [CrossRef]
- Sortais, J.-B.; Pannetier, N.; Holuigue, A.; Barloy, L.; Sirlin, C.; Pfeffer, M.; Kyritsakas, N. Cyclometalation of Primary Benzyl Amines by Ruthenium(II), Rhodium(III), and Iridium(III) Complexes. Organometallics 2007, 26, 1856–1867. [Google Scholar] [CrossRef]
- Sortais, J.-B.; Pannetier, N.; Clément, N.; Barloy, L.; Sirlin, C.; Pfeffer, M.; Kyritsakas, N. Cyclometallation of Secondary Benzyl Amines by Ruthenium(II) Complexes. Organometallics 2007, 26, 1868–1874. [Google Scholar] [CrossRef]
- Arita, S.; Koike, T.; Kayaki, Y.; Ikariya, T. Synthesis and Reactivities of Cp*Ir Amide and Hydride Complexes Bearing C−N Chelate Ligands. Organometallics 2008, 27, 2795–2802. [Google Scholar] [CrossRef]
- Arita, S.; Koike, T.; Kayaki, Y.; Ikariya, T. Aerobic Oxidation of Alcohols with Bifunctional Transition-Metal Catalysts Bearing C–N Chelate Ligands. Chem. Asian J. 2008, 3, 1479–1485. [Google Scholar] [CrossRef]
- Sortais, J.-B.; Ritleng, V.; Voelklin, A.; Holuigue, A.; Smail, H.; Barloy, L.; Sirlin, C.; Verzijl, G.K.M.; Boogers, J.A.F.; de Vries, A.H.M.; et al. Cycloruthenated Primary and Secondary Amines as Efficient Catalyst Precursors for Asymmetric Transfer Hydrogenation. Org. Lett. 2005, 7, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Sortais, J.B.; Barloy, L.; Sirlin, C.; de Vries, A.H.M.; de Vries, J.G.; Pfeffer, M. Cycloruthenated compounds as efficient catalyst for asymmetric hydride transfer reaction. Pure Appl. Chem. 2006, 78, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Pannetier, N.; Sortais, J.-B.; Dieng, P.S.; Barloy, L.; Sirlin, C.; Pfeffer, M.; Kyritsakas, N. Kinetics and Mechanism of Ruthenacycle-Catalyzed Asymmetric Hydrogen Transfer. Organometallics 2008, 27, 5852–5879. [Google Scholar] [CrossRef]
- Jerphagnon, T.; Haak, R.; Berthiol, F.; Gayet, A.J.A.; Ritleng, V.; Holuigue, A.; Pannetier, N.; Pfeffer, M.; Voelklin, A.; Lefort, L.; et al. Ruthenacycles and Iridacycles as Catalysts for Asymmetric Transfer Hydrogenation and Racemisation. Top. Catal. 2010, 53, 1002–1008. [Google Scholar] [CrossRef] [Green Version]
- Barloy, L.; Issenhuth, J.-T.; Weaver, M.G.; Pannetier, N.; Sirlin, C.; Pfeffer, M. Reaction of Chiral Secondary Amines with [(η5-C5Me5)MCl2]2 (M = Rh(III), Ir(III)): Cyclometalation with or without Dehydrogenation. Organometallics 2011, 30, 1168–1174. [Google Scholar] [CrossRef]
- Pannetier, N.; Sortais, J.-B.; Issenhuth, J.-T.; Barloy, L.; Sirlin, C.; Holuigue, A.; Lefort, L.; Panella, L.; de Vries, J.G.; Pfeffer, M. Cyclometalated Complexes of Ruthenium, Rhodium and Iridium as Catalysts for Transfer Hydrogenation of Ketones and Imines. Adv. Synth. Catal. 2011, 353, 2844–2852. [Google Scholar] [CrossRef] [Green Version]
- Féghali, E.; Barloy, L.; Issenhuth, J.-T.; Karmazin-Brelot, L.; Bailly, C.; Pfeffer, M. Cyclometalation of (2R,5R)-2,5-Diphenylpyrrolidine and 2- Phenyl-2-imidazoline Ligands with Half-Sandwich Iridium(III) and Rhodium(III) Complexes. Organometallics 2013, 32, 6186–6194. [Google Scholar] [CrossRef]
- Sato, Y.; Kayaki, Y.; Ikariya, T. Efficient dynamic kinetic resolution of racemic secondary alcohols by a chemoenzymatic system using bifunctional iridium complexes with C–N chelate amido ligands. Chem. Commun. 2012, 48, 3635–3637. [Google Scholar] [CrossRef] [PubMed]
- Ueno, A.; Kayaki, Y.; Ikariya, T. Heterolysis of NH-Indoles by Bifunctional Amido Complexes and Applications to Carboxylation with Carbon Dioxide. Organometallics 2014, 33, 4479–4485. [Google Scholar] [CrossRef]
- Sato, Y.; Kayaki, Y.; Ikariya, T. Cationic Iridium and Rhodium Complexes with C–N Chelating Primary Benzylic Amine Ligands as Potent Catalysts for Hydrogenation of Unsaturated Carbon–Nitrogen Bonds. Organometallics 2016, 35, 1257–1264. [Google Scholar] [CrossRef]
- Matsunami, A.; Kuwata, S.; Kayaki, Y. Hydrodefluorination of Fluoroarenes Using Hydrogen Transfer Catalysts with a Bifunctional Iridium/NH Moiety. ACS Catal. 2016, 6, 5181–5185. [Google Scholar] [CrossRef]
- Sato, Y.; Kayaki, Y.; Ikariya, T. Comparative Study of Bifunctional Mononuclear and Dinuclear Amidoiridium Complexes with Chiral C−N Chelating Ligands for the Asymmetric Transfer Hydrogenation of Ketones. Chem. Asian J. 2016, 11, 2924–2931. [Google Scholar] [CrossRef]
- Sato, Y.; Kayaki, Y.; Ikariya, T. Transfer hydrogenation of carbon dioxide via bicarbonate promoted by bifunctional C–N chelating Cp*Ir(III) complexes. Chem. Commun. 2020, 56, 10762–10765. [Google Scholar] [CrossRef]
- Tsuboyama, A.; Takiguchi, T.; Okada, S.; Osawa, M.; Hoshino, M.; Ueno, K. A novel dinuclear cyclometalated iridium complex bridged with 1,4-bis[pyridine-2-yl]benzene: Its structure and photophysical properties. Dalton Trans. 2004, 1115–1116. [Google Scholar] [CrossRef]
- Ho, J.H.H.; Choy, S.W.S.; Macgregor, S.A.; Messerle, B.A. Cooperativity in Bimetallic Dihydroalkoxylation Catalysts Built on Aromatic Scaffolds: Significant Rate Enhancements with a Rigid Anthracene Scaffold. Organometallics 2011, 30, 5978–5984. [Google Scholar] [CrossRef]
- Choy, S.W.S.; Page, M.J.; Bhadbhade, M.; Messerle, B.A. Cooperative Catalysis: Large Rate Enhancements with Bimetallic Rhodium Complexes. Organometallics 2013, 32, 4726–4729. [Google Scholar] [CrossRef]
- Maity, R.; Hohloch, A.; Su, C.-Y.; Meer, M.; Sarker, B. Cyclometalated Mono- and Dinuclear IrIII Complexes with “Click”-Derived Triazoles and Mesoionic Carbenes. Chem. Eur. J. 2014, 20, 9952–9961. [Google Scholar] [CrossRef] [PubMed]
- Hull, J.F.; Himeda, Y.; Wang, W.-H.; Hashiguchi, B.; Periana, R.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat. Chem. 2012, 4, 383–388. [Google Scholar] [CrossRef]
- Wang, Y.; Kuzniewski, C.N.; Rauniyar, V.; Hoong, C.; Toste, F.D. Chiral (Acyclic Diaminocarbene) Gold(I)-Catalyzed Dynamic Kinetic Asymmetric Transformation of Propargyl Esters. J. Am. Chem. Soc. 2011, 133, 12972–12975. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, S.; Katayama, T.; Onitsuka, K.; Suzuki, T.; Yanagida, T.; Kawai, T.; Sasai, H. Chiral dinuclear vanadium(v) catalysts for oxidative coupling of 2-naphthols. Chem. Commun. 2008, 44, 1810–1812. [Google Scholar] [CrossRef]
- Sako, M.; Aoki, T.; Zumbrägel, N.; Schober, L.; Gröger, H.; Takizawa, S.; Sasai, H. Chiral Dinuclear Vanadium Complex-Mediated Oxidative Coupling of Resorcinols. J. Org. Chem. 2019, 84, 1580–1587. [Google Scholar] [CrossRef]
- Shi, M.; Itoh, N.; Masaki, Y. Axially Dissymmetric Chiral (Diamine) copper- and (Diimine)copper-catalysed Asymmetric Aziridination of Alkenes. J. Chem. Res. (S) 1996, 8, 352–353. [Google Scholar]
- Matsumoto, K.; Tomioka, K. An approach to a chiral cycloalkanone-mediated asymmetric epoxidation of stilbene with oxone. Chem. Pharm. Bull. 2001, 49, 1653–1657. [Google Scholar] [CrossRef] [Green Version]
- Takagi, K.; Okamoto, T.; Sakakibara, Y.; Oka, S. PALLADIUM(II) CATALYZED SYNTHESIS OF ARYL CYANIDES FROM ARYL HALIDES. Chem. Lett. 1973, 2, 471. [Google Scholar] [CrossRef]
- Sakakibara, Y.; Okuda, F.; Shimobayashi, A.; Kirino, K.; Sakai, M.; Uchida, N.; Takagi, K. The Cyanation of Aromatic Halides Catalyzed by Nickel(0) Complexes Generated In Situ. I. General Scope and Limitations. Bull. Chem. Soc. Jpn. 1988, 61, 1985. [Google Scholar] [CrossRef] [Green Version]
- Takagi, K.; Sakakibara, Y. Nickel(0) or Palladium(0)-catalyzed Cyanation of Aryl Triflates. Chem. Lett. 1989, 18, 1957–1958. [Google Scholar] [CrossRef]
- Takagi, K.; Sakai, K.; Sakakibara, Y. Nucleophilic Displacement Catalyzed by Transition Metal. IX. [Pd2(dba)3]·CHCl3–DPPF Catalyzed Cyanation of Aryl Halides and Ary Triflates. Bull. Chem. Soc. Jpn. 1991, 64, 1118. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, Y.; Sasaki, K.; Okuda, F.; Hokimoto, A.; Ueda, T.; Sakai, M.; Takagi, K. The Cyanation of Aromatic Halides Catalyzed by Nickel(0) Complexes Generated In Situ. III. Kinetic and Mechanistic Aspects. Bull. Chem. Soc. Jpn. 2004, 77, 1013–1019. [Google Scholar] [CrossRef]
- Sundermeier, M.; Zapf, A.; Beller, M. Palladium--Catalyzed Cyanation of Aryl Halides: Recent Developments and Perspectives. Eur. J. Inorg. Chem. 2003, 3513–3526. [Google Scholar] [CrossRef]
- Schareina, T.; Jackstell, R.; Schulz, T.; Zapf, A.; Cotté, A.; Gotta, M.; Beller, M. Increasing the Scope of Palladium--Catalyzed Cyanations of Aryl Chlorides. Adv. Synth. Catal. 2009, 351, 643–648. [Google Scholar] [CrossRef]
- Anderson, B.A.; Bell, E.C.; Ginah, F.O.; Harn, N.K.; Pagh, L.M.; Wepsiec, J.P. Cooperative Catalyst Effects in Palladium-Mediated Cyanation Reactions of Aryl Halides and Triflates. J. Org. Chem. 1998, 63, 8224–8228. [Google Scholar] [CrossRef]
- Buono, F.G.; Chidambaram, R.; Mueller, R.H.; Waltermire, R.E. Insights into palladium-catalyzed cyanation of bromobenzene: Additive effects on the rate-limiting step. Org. Lett. 2008, 10, 5325–5328. [Google Scholar] [CrossRef] [PubMed]
- Kurz, L.; Lee, G.; Morgans, D., Jr.; Waldyke, M.J.; Ward, T. Stereospecific functionalization of (R)-(−)-1,1′-bi-2-naphthol triflate. Tetrahedron Lett. 1990, 31, 6321. [Google Scholar]
- Kasak, P.; Putala, M. Stereoconservative cyanation of [1,1′-binaphthalene]-2,2′-dielectrophiles. An alternative approach to homochiral C2-symmetric [1,1′-binaphthalene]-2,2′-dicarbonitrile and its transformations. Collect. Czech. Chem. Commun. 2000, 65, 729–740. [Google Scholar] [CrossRef]
- Wu, B.; Zhang, J.; Yang, M.; Ma, L.-J.; Yu, X.-Q. Raney Ni/KBH4: An efficient and mild system for the reduction of nitriles to amines. ARKIVOC 2008, 12, 95–102. [Google Scholar] [CrossRef] [Green Version]
- White, C.; Yates, A.; Maitlis, P.M.; Heinekey, D.M. (η5-Pentamethylcyclopentadienyl)rhodium and -iridium compounds. Inorg. Synth. 1992, 29, 228–234. [Google Scholar]
- Booth, G.; Chatt, J. Some Complexes of Ditertiary Phosphines with Nickel(II) and Nickel (III). J. Chem. Soc. 1965, 3238–3241. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
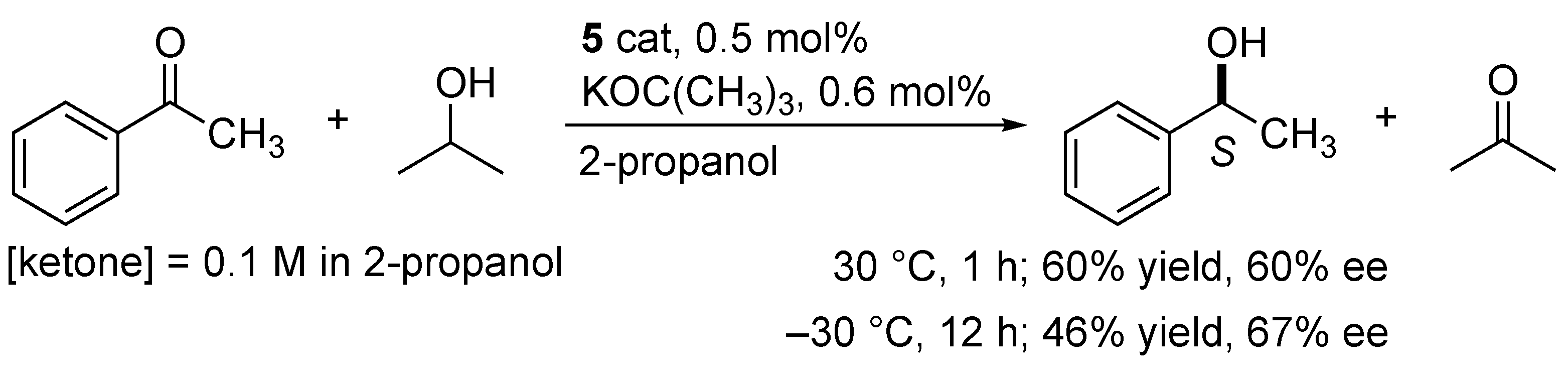{kind=link}
| Entry | Cat | Ligand | Solvent | % Yield 1 | |
|---|---|---|---|---|---|
| 3 | 4 | ||||
| 1 | NiCl2(dppe) | DPPE | Dioxane | 2 | n.d. 2 |
| 2 | NiCl2(dppe) | DPPE | CH3CN | 19 | 17 |
| 3 | NiCl2(dppe) | DPPE | DMF | 87 | 0 |
| 4 | NiCl2(dppp) | DPPP | DMF | 57 | 5 |
| 5 | NiCl2(dppf) | DPPF | DMF | 17 | n.d. 2 |
| 6 | PdCl2(dppe) | DPPE | DMF | 59 | n.d. 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sato, Y.; Kawata, Y.; Yasui, S.; Kayaki, Y.; Ikariya, T. New Bifunctional Bis(azairidacycle) with Axial Chirality via Double Cyclometalation of 2,2′-Bis(aminomethyl)-1,1′-binaphthyl. Molecules 2021, 26, 1165. https://doi.org/10.3390/molecules26041165
Sato Y, Kawata Y, Yasui S, Kayaki Y, Ikariya T. New Bifunctional Bis(azairidacycle) with Axial Chirality via Double Cyclometalation of 2,2′-Bis(aminomethyl)-1,1′-binaphthyl. Molecules. 2021; 26(4):1165. https://doi.org/10.3390/molecules26041165
Chicago/Turabian StyleSato, Yasuhiro, Yuichi Kawata, Shungo Yasui, Yoshihito Kayaki, and Takao Ikariya. 2021. "New Bifunctional Bis(azairidacycle) with Axial Chirality via Double Cyclometalation of 2,2′-Bis(aminomethyl)-1,1′-binaphthyl" Molecules 26, no. 4: 1165. https://doi.org/10.3390/molecules26041165
APA StyleSato, Y., Kawata, Y., Yasui, S., Kayaki, Y., & Ikariya, T. (2021). New Bifunctional Bis(azairidacycle) with Axial Chirality via Double Cyclometalation of 2,2′-Bis(aminomethyl)-1,1′-binaphthyl. Molecules, 26(4), 1165. https://doi.org/10.3390/molecules26041165