
Point Mutations of Nicotinic Receptor α1 Subunit Reveal New Molecular Features of G153S Slow-Channel Myasthenia

 , ,
, ,
Abstract
:
1. Introduction
2. Results
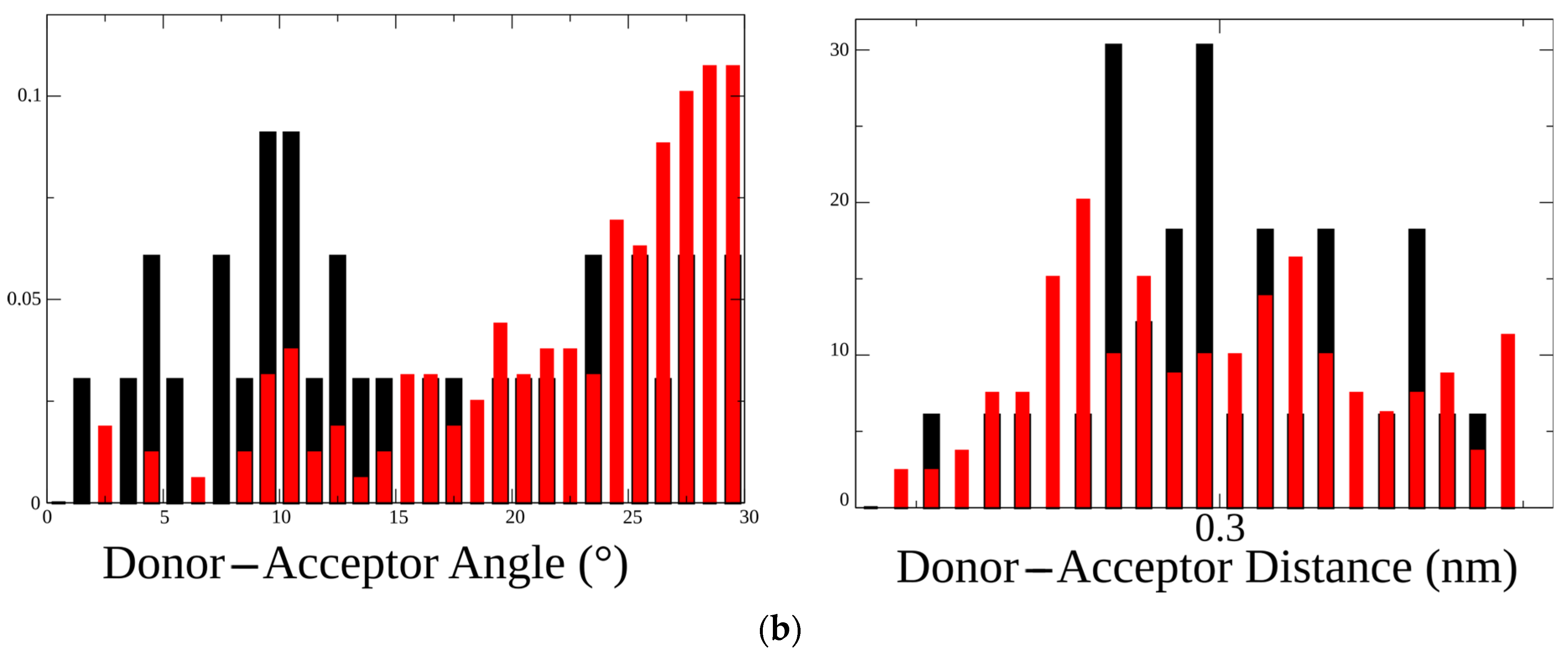
2.1. Molecular Dynamics of Extracellular Domain in α1(G153S) Mutant
2.2. Design of Double Mutants to Modify H-Bond Arrangement of 153-rd Residue
2.3. Fluorescent Ca2+ Assay of Mutated Muscle nAChRs
2.4. Patch-Clamp Investigation of WT and G153S Muscle nAChRs Functional Properties
3. Discussion
4. Materials and Methods
4.1. Molecular Modeling
4.2. Point Mutagenesis and Transient Transfection
4.3. Calcium Fluorescent Imaging
4.4. Electrophysiology
4.5. Data Analisys
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cetin, H.; Beeson, D.; Vincent, A.; Webster, R. The structure, function, and physiology of the fetal and adult acetylcholine receptor in muscle. Front. Mol. Neurosci. 2020, 13, 170. [Google Scholar] [CrossRef] [PubMed]
- Fichtner, M.L.; Jiang, R.; Bourke, A.; Nowak, R.J.; O’Connor, K.C. Autoimmune pathology in myasthenia gravis disease subtypes is governed by divergent mechanisms of immunopathology. Front. Immunol. 2020, 11, 776. [Google Scholar] [CrossRef]
- Engel, A.G. Congenital myasthenic syndromes in 2018. Curr. Neurol. Neurosci. Rep. 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhaesebrouck, A.E.; Beeson, D. The congenital myasthenic syndromes. Curr. Opin. Neurol. 2019, 32, 696–703. [Google Scholar] [CrossRef]
- Mignan, T.; Targett, M.; Lowrie, M. Classification of myasthenia gravis and congenital myasthenic syndromes in dogs and cats. J. Vet. Intern. Med. 2020, 34, 1707–1717. [Google Scholar] [CrossRef]
- Kudryavtsev, D.S.; Spirova, E.N.; Shelukhina, I.V.; Son, L.V.; Makarova, Y.V.; Utkina, N.K.; Kasheverov, I.E.; Tsetlin, V.I. Makaluvamine G from the marine sponge zyzzia fuliginosa inhibits muscle nAChR by binding at the orthosteric and allosteric sites. Mar. Drugs 2018, 16, 109. [Google Scholar] [CrossRef] [Green Version]
- Shelukhina, I.; Spirova, E.; Kudryavtsev, D.; Ojomoko, L.; Werner, M.; Methfessel, C.; Hollmann, M.; Tsetlin, V. Calcium imaging with genetically encoded sensor Case12: Facile analysis of α7/α9 nAChR mutants. PLoS ONE 2017. [Google Scholar] [CrossRef] [Green Version]
- Engel, A.G.; Lambert, E.H.; Mulder, D.M.; Torres, C.F.; Sahashi, K.; Bertorini, T.E.; Whitaker, J.N. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann. Neurol. 1982, 11, 553–569. [Google Scholar] [CrossRef]
- Bouzat, C. Ephedrine blocks wild-type and long-lived mutant acetylcholine receptor channels. Neuroreport 1996, 8, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Cruz, P.M.; Palace, J.; Ramjattan, H.; Jayawant, S.; Robb, S.A.; Beeson, D. Salbutamol and ephedrine in the treatment of severe AChR deficiency syndromes. Neurology 2015, 85, 1043–1047. [Google Scholar] [CrossRef] [Green Version]
- Fukudome, T.; Ohno, K.; Brengman, J.M.; Engel, A.G. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. Neuroreport 1998, 9, 1907–1911. [Google Scholar] [CrossRef]
- Gisselmann, G.; Alisch, D.; Welbers-Joop, B.; Hatt, H. Effects of quinine, quinidine and chloroquine on human muscle nicotinic acetylcholine receptors. Front. Pharmacol. 2018, 9, 1339. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.M.; Fukodome, T.; Engel, A.G. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology 2003, 60, 1710–1713. [Google Scholar] [CrossRef] [PubMed]
- Sine, S.M.; Ohno, K.; Bouzat, C.; Auerbach, A.; Milone, M.; Pruitt, J.N.; Engel, A.G. Mutation of the acetylcholine receptor α subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity. Neuron 1995, 15, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Iyadurai, S.J.P. Congenital myasthenic syndromes. Neurol. Clin. 2020, 38, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Puskar, N.L.; Lester, H.A.; Dougherty, D.A. Probing the effects of residues located outside the agonist binding site on drug-receptor selectivity in the nicotinic receptor. ACS Chem. Biol. 2012, 7, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Grassi, F.; Fucile, S. Calcium influx through muscle nAChR-channels: One route, multiple roles. Neuroscience 2020, 439, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.M.; Maselli, R.A.; Groshong, J.; Zayas, R.; Wollmann, R.L.; Cens, T.; Charnet, P. Active calcium accumulation underlies severe weakness in a panel of mice with slow-channel syndrome. J. Neurosci. 2002, 22, 6447–6457. [Google Scholar] [CrossRef]
- Webster, R.G.; Cossins, J.; Lashley, D.; Maxwell, S.; Liu, W.W.; Wickens, J.R.; Martinez-Martinez, P.; de Baets, M.; Beeson, D. A mouse model of the slow channel myasthenic syndrome: Neuromuscular physiology and effects of ephedrine treatment. Exp. Neurol. 2013, 248, 286–298. [Google Scholar] [CrossRef]
- Rahman, M.M.; Teng, J.; Worrell, B.T.; Noviello, C.M.; Lee, M.; Karlin, A.; Stowell, M.H.B.; Hibbs, R.E. Structure of the native muscle-type nicotinic receptor and inhibition by snake venom toxins. Neuron 2020, 106, 962. [Google Scholar] [CrossRef]
- Zdenek, C.N.; Harris, R.J.; Kuruppu, S.; Youngman, N.J.; Dobson, J.S.; Debono, J.; Khan, M.; Smith, I.; Yarski, M.; Harrich, D.; et al. A taxon-specific and high-throughput method for measuring ligand binding to nicotinic acetylcholine receptors. Toxins 2019, 11, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryavtsev, D.S.; Tabakmakher, V.; Budylin, G.S.; Egorova, N.S.; Efremov, R.G.; Ivanov, I.A.; Belukhina, S.Y.; Jegorov, A.V.; Kasheverov, I.E.; Kryukova, E.V.; et al. Complex approach for analysis of snake venom α-neurotoxins binding to HAP, the high-affinity peptide. Sci. Rep. 2020, 10, 3816. [Google Scholar] [CrossRef]
- Lynagh, T.; Kiontke, S.; Meyhoff-Madsen, M.; Gless, B.H.; Johannesen, J.; Kattelmann, S.; Christiansen, A.; Dufva, M.; Laustsen, A.H.; Devkota, K.; et al. Peptide inhibitors of the α-cobratoxin-nicotinic acetylcholine receptor interaction. J. Med. Chem. 2020, 63, 13709–13718. [Google Scholar] [CrossRef]
- Finkelstein, A.V.; Ptitsyn, O.B. Protein Physics: A Course of Lectures; Elsevier Ltd.: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Eswar, N.; Ramakrishnan, C. Deterministic features of side-chain main-chain hydrogen bonds in globular protein structures. Protein Eng. Des. Sel. 2000, 13, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, S.A.; Firestone, L.L.; Miller, K.W. Is agonist self-inhibition at the nicotinic acetylcholine receptor a nonspecific action? Biochemistry 1987, 26, 2807–2814. [Google Scholar] [CrossRef] [PubMed]
- Arias, H.R. Agonist self-inhibitory binding site of the nicotinic acetylcholine receptor. J. Neurosci. Res. 1996, 44, 97–105. [Google Scholar] [CrossRef]
- Gomez, C.M.; Maselli, R.; Gammack, J.; Lasalde, J.; Tamamizu, S.; Cornblath, D.R.; Lehar, M.; McNamee, M.; Kuncl, R.W. A β-subunit mutation in the acetylcholine receptor channel gate causes severe slow-channel syndrome. Ann. Neurol. 1996, 39, 712–723. [Google Scholar] [CrossRef]
- Otero-Cruz, J.D.; Báez-Pagán, C.A.; Dorna-Pérez, L.; Grajales-Reyes, G.E.; Ramírez-Ordoñez, R.T.; Luciano, C.A.; Gómez, C.M.; Lasalde-Dominicci, J.A. Decoding pathogenesis of slow-channel congenital myasthenic syndromes using recombinant expression and mice models. NIH Public Access 2010, 29, 4–17. [Google Scholar]
- Tripathy, S.; Zheng, W.; Auerbach, A. A single molecular distance predicts agonist binding energy in nicotinic receptors. J. Gen. Physiol. 2019, 151, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Chevessier, F.F.; Peter, C.; Mersdorf, U.; Girard, E.; Krejci, E.; McArdle, J.J.; Witzemann, V. A new mouse model for the slow-channel congenital myasthenic syndrome induced by the AChR εL221F mutation. Neurobiol. Dis. 2012, 45, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.G.; Sikorska, M.; Sandhu, J.K.; Lanthier, P.; Ribecco-Lutkiewicz, M.; Bani-Yaghoub, M. Differentiation of mouse Neuro 2A cells into dopamine neurons. J. Neurosci. Methods 2010, 186, 60–67. [Google Scholar] [CrossRef]
- Mao, A.J.; Bechberger, J.; Lidington, D.; Galipeau, J.; Laird, D.W.; Naus, C.C.G. Neuronal differentiation and growth control of neuro-2a cells after retroviral gene delivery of connexin43. J. Biol. Chem. 2000, 275, 34407–34414. [Google Scholar] [CrossRef] [Green Version]
- Aidoo, A.Y.; Ward, K. Spatio-temporal concentration of acetylcholine in vertebrate synaptic cleft. Math. Comput. Model. 2006, 44, 952–962. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapovalov, M.V.; Dunbrack, R.L. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Gu, S.; Matta, J.A.; Lord, B.; Harrington, A.W.; Sutton, S.W.; Davini, W.B.; Bredt, D.S. Brain α7 nicotinic acetylcholine receptor assembly requires NACHO. Neuron 2016, 89, 948–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halevi, S.; Yassin, L.; Eshel, M.; Sala, F.; Sala, S.; Criado, M.; Treinin, M. Conservation within the RIC-3 gene family: Effectors of mammalian nicotinic acetylcholine receptor expression. J. Biol. Chem. 2003, 278, 34411–34417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshpande, A.; Vinayakamoorthy, R.M.; Garg, B.K.; Thummapudi, J.P.; Oza, G.; Adhikari, K.; Agarwal, A.; Dalvi, P.; Iyer, S.; Thulasi Raman, S.; et al. Why does knocking out NACHO, but not RIC3, completely block expression of α7 nicotinic receptors in mouse brain? Biomolecules 2020, 10, 470. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Fukuda, K.; Konno, T.; Mori, Y.; Tanaka, K.I.; Mishina, M.; Numa, S. Functional properties of nicotinic acetylcholine receptor subunits expressed in various combinations. FEBS Lett. 1987, 214, 253–258. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | EC50 | Time Constant, s |
|---|---|---|
| WT | 2503 nM (2268 nM, 2763 nM) 1 | 15.0 (14.6, 15.4) |
| L199A | 1035 nM (488 nM, 2199 nM) | 19.2 (18.9, 19.5) |
| L199T | 2607 nM (2341 nM, 2903 nM) | 14.2 (13.9, 14.5) |
| L199A/G153S | 491 nM (176 nM, 1369 nM) | 14.3 (14.0, 14.6) |
| L199T/G153S | 579 nM (392 nM, 856 nM) | 11.1 (10.8, 11.4) |
| G153S | 146 nM (122 nM, 174 nM) | 34.0 (33.6, 34.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudryavtsev, D.; Isaeva, A.; Barkova, D.; Spirova, E.; Mukhutdinova, R.; Kasheverov, I.; Tsetlin, V. Point Mutations of Nicotinic Receptor α1 Subunit Reveal New Molecular Features of G153S Slow-Channel Myasthenia. Molecules 2021, 26, 1278. https://doi.org/10.3390/molecules26051278
Kudryavtsev D, Isaeva A, Barkova D, Spirova E, Mukhutdinova R, Kasheverov I, Tsetlin V. Point Mutations of Nicotinic Receptor α1 Subunit Reveal New Molecular Features of G153S Slow-Channel Myasthenia. Molecules. 2021; 26(5):1278. https://doi.org/10.3390/molecules26051278
Chicago/Turabian StyleKudryavtsev, Denis, Anastasia Isaeva, Daria Barkova, Ekaterina Spirova, Renata Mukhutdinova, Igor Kasheverov, and Victor Tsetlin. 2021. "Point Mutations of Nicotinic Receptor α1 Subunit Reveal New Molecular Features of G153S Slow-Channel Myasthenia" Molecules 26, no. 5: 1278. https://doi.org/10.3390/molecules26051278
APA StyleKudryavtsev, D., Isaeva, A., Barkova, D., Spirova, E., Mukhutdinova, R., Kasheverov, I., & Tsetlin, V. (2021). Point Mutations of Nicotinic Receptor α1 Subunit Reveal New Molecular Features of G153S Slow-Channel Myasthenia. Molecules, 26(5), 1278. https://doi.org/10.3390/molecules26051278