
Semi-Synthesis of Harringtonolide Derivatives and Their Antiproliferative Activity


Abstract
:1. Introduction
2. Results
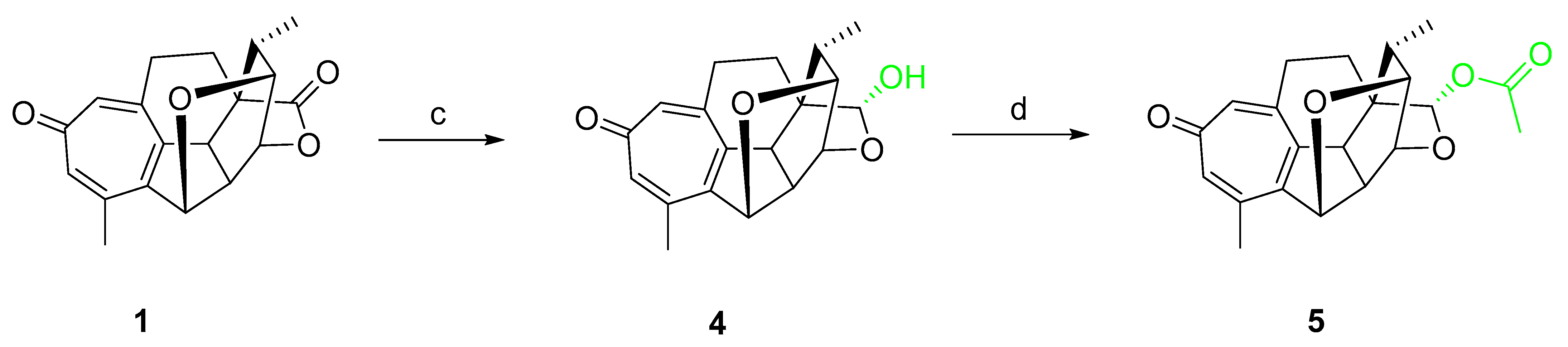
2.1. Chemical Synthesis
2.2. Cytotoxicity
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of Compounds 2 and 3
4.1.2. General Procedure for the Synthesis of Compounds 4 and 5
4.1.3. General Procedure for the Synthesis of Compounds 6 and 7
4.1.4. General Procedure for the Synthesis of Compounds 8–13
4.2. Cell Culture
4.3. MTT Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- He, Y.R.; Shen, Y.H.; Shan, L.; Yang, X.; Wen, B.; Ye, J.; Yuan, X.; Li, H.L.; Xu, X.K.; Zhang, W.D. Diterpenoid Lanceolatins A–G from Cephalotaxus Lanceolata and Their Anti-Inflammatory and Anti-Tumor Activities. RSC Adv. 2015, 5, 4126–4134. [Google Scholar] [CrossRef]
- Huang, Q.; Guo, K.; Liu, Y.; Liu, Y.; Li, W.; Geng, H.; Wang, Y.; Li, S. Diterpenoids and Flavonoids from the Twigs of Cephalotaxus Fortunei Var. Alpina. Chem. Biodivers 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.X.; Fan, Y.Y.; Xu, J.B.; Gan, L.S.; Xu, C.H.; Ding, J.; Yue, J.M. Diterpenoids and Lignans from Cephalotaxus Fortunei. J. Nat. Prod. 2017, 80, 356–362. [Google Scholar] [CrossRef]
- Zhao, C.X.; Li, B.Q.; Shao, Z.X.; Li, D.H.; Jing, Y.K.; Li, Z.L.; Hua, H.M. Cephasinenoside A, a New Cephalotane Diterpenoid Glucoside from Cephalotaxus Sinensis. Tetrahedron Lett. 2019, 60, 151154. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Shao, Z.; Zhao, C.; Jing, Q.; Li, D.; Lin, B.; Jing, Y.; Li, Z.; Hua, H. Diterpenoids from Cephalotaxus Fortunei Var. Alpina and Their Cytotoxic Activity. Bioorg.c Chem. 2020, 103, 104226. [Google Scholar] [CrossRef]
- Buta, J.G.; Flippen, J.L.; Lusby, W.R. Harringtonolide, a Plant Growth Inhibitory Tropone from Cephalotaxus Harringtonia (Forbes) K. Koch. J. Org. Chem. 1978, 43, 1002–1003. [Google Scholar] [CrossRef]
- Kang, S.Q.; Cai, S.Y.; Teng, L. The Antiviral Effect of Hainanolide, A Compound Isolated from Cephaloyaxis Hainanensis. Acta Pharm Sin. 1981, 16, 867–868. [Google Scholar]
- Ni, L.; Zhong, X.H.; Chen, X.J.; Zhang, B.J.; Bao, M.F.; Cai, X.H. Bioactive Norditerpenoids from Cephalotaxus Fortunei Var. Alpina and C. Lanceolata. Phytochemistry 2018, 151, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Evanno, L.; Jossang, A.; Pouplin, J.N.; Delaroche, D.; Herson, P.; Seuleiman, M.; Bodo, B.; Nay, B. Further Studies of the Norditerpene (+)-Harringtonolide Isolated from Cephalotaxus Harringtonia Var. Drupacea: Absolute Configuration, Cytotoxic and Antifungal Activities. Planta. Med. 2008, 74, 870–872. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.J.; Hu, L.; Ma, Z.; Li, R.; Zhang, Z.; Tao, C.; Cheng, B.; Li, Y.; Wang, H.; Zhai, H. Total Synthesis of the Diterpenoid (+)-Harringtonolide. Angew. Chem. 2016, 128, 11810–11813. [Google Scholar] [CrossRef]
- Ma, Z.; Cheng, B.; Zhai, H. Synthetic Studies Toward Harringtonolide. Asian J. Org. Chem. 2014, 3, 1097–1101. [Google Scholar] [CrossRef]
- Abdelkafi, H.; Herson, P.; Nay, B. Asymmetric Synthesis of the Oxygenated Polycyclic System of (+)-Harringtonolide. Org. Lett. 2012, 14, 1270–1273. [Google Scholar] [CrossRef]
- O’Sullivan, T.P.; Zhang, H.; Mander, L.N. Model Studies toward the Synthesis of the Bioactive Diterpenoid, Harringtonolide. Org. Biomol. Chem. 2007, 5, 2627. [Google Scholar] [CrossRef] [Green Version]
- Frey, B.; Wells, A.P.; Rogers, D.H.; Mander, L.N. Synthesis of the Unusual Diterpenoid Tropones Hainanolidol and Harringtonolide. J. Am. Chem. Soc. 1998, 120, 1914–1915. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, N.; Tang, W. Stereoselective Total Synthesis of Hainanolidol and Harringtonolide via Oxidopyrylium-Based [5 + 2] Cycloaddition. J. Am. Chem. Soc. 2013, 135, 12434–12438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.B.; Appels, D.C.; Hockless, D.C.R.; Mander, L.N. A New Approach to the Total Synthesis of the Unusual Diterpenoid Tropone, Harringtonolide. Tetrahedron Lett. 1998, 39, 6577–6580. [Google Scholar] [CrossRef]
- Abdelkafi, H.; Evanno, L.; Herson, P.; Nay, B. Synthetic Studies toward the Cytotoxic Norditerpene (+)-Harringtonolide: Setting up Key-Stereogenic Centers of the Cyclohexane Ring D. Tetrahedron Lett. 2011, 52, 3447–3450. [Google Scholar] [CrossRef]
- Frey, B.; Mander, L.N.; Hockless, D.C.R. Ether Formation by Intramolecular Attack of Hydroxy on Cyclopropyl Rings: A Model for the Formation of the Tetrahydrofuran Moiety in the Diterpenoid, Harringtonolide. J. Chem. Soc. Perkin Trans. 1 1998, 1555–1560. [Google Scholar] [CrossRef]
- Sun, N.J.; Xue, Z.; Liang, X.T.; Huang, L. Studies on the Structure of A New Antitumor Agent-Hainanolide. Acta Pharm. Sin. 1979, 14, 41–46. [Google Scholar]
- Fan, Y.Y.; Xu, J.B.; Liu, H.C.; Gan, L.S.; Ding, J.; Yue, J.M. Cephanolides A–J, Cephalotane-Type Diterpenoids from Cephalotaxus Sinensis. J. Nat. Prod. 2017, 80, 3159–3166. [Google Scholar] [CrossRef]
- Ge, Z.P.; Liu, H.C.; Wang, G.C.; Liu, Q.F.; Xu, C.F.; Xu, C.F.; Ding, J.; Fan, Y.Y.; Yue, J.M. 17-nor-Cephalotane-Type Diterpenoids from Cephalotaxus Fortunei. J. Nat. Prod. 2019, 82, 1565–1575. [Google Scholar] [CrossRef]
- Du, J.; Chiu, M.H.; Nie, R.L. Two New Lactones from Cephalotaxus Fortunei Var. Alpnia. J. Nat. Prod. 1999, 62, 1664–1665. [Google Scholar] [CrossRef]
- Ni, G.; Zhang, H.; Fan, Y.Y.; Liu, H.C.; Ding, J.; Yue, J.M. Mannolides A–C with an Intact Diterpenoid Skeleton Providing Insights on the Biosynthesis of Antitumor Cephalotaxus Troponoids. Org. Lett. 2016, 18, 1880–1883. [Google Scholar] [CrossRef]
- Wu, G.R.; Xu, B.; Yang, Y.Q.; Zhang, X.Y.; Fang, K.; Ma, T.; Wang, H.; Xue, N.N.; Chen, M.; Guo, W.B.; et al. Synthesis and Bio-logical Evaluation of Podophyllotoxin Derivatives as Selective Antitumor Agents. Eur. J. Med. Chem. 2018, 155, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Stern, P.H.; Hoffman, R.M. Enhanced In Vitro Selective Toxicity of Chemotherapeutic Agents for Human Cancer Cells Based on a Metabolic Defect. J. Natl. Cancer Inst. 1986, 76, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Haglund, C.; Åleskog, A.; Nygren, P.; Gullbo, J.; Höglund, M.; Wickström, M.; Larsson, R.; Lindhagen, E. In Vitro Evaluation of Clinical Activity and Toxicity of Anticancer Drugs Using Tumor Cells from Patients and Cells Representing Normal Tissues. Cancer Chemother. Pharmacol. 2012, 69, 697–707. [Google Scholar] [CrossRef]
- You, Y.J.; Kim, Y.; Nam, N.H.; Ahn, B.Z. Synthesis and Cytotoxic Activity of A-Ring Modified Betulinic Acid Derivatives. Bioorg. Med. Chem. Lett. 2003, 13, 3137–3140. [Google Scholar] [CrossRef]
- Rao, Z.; Liu, X.; Zhou, W.; Yi, J.; Li, S.S. Synthesis and Antitumour Activity of β-Hydroxyisovalerylshikonin Analogues. Eur. J. Med. Chem. 2011, 46, 3934–3941. [Google Scholar] [CrossRef]
- Demidov, M.R.; Lapshina, M.Y.; Osipov, D.V.; Osyanin, V.A.; Klimochkin, Y.N. Oxidative Rearrangement of 4H-Chromenes to 2-Aroylbenzofurans in the Presence of Selenium Dioxide. Chem. Heterocycl. Comp. 2017, 53, 1053–1056. [Google Scholar] [CrossRef]
- Morita, S.; Yoshimura, T.; Matsuo, J. Intramolecular Büchner Reaction and Oxidative Aromatization with SeO2 or O2. Chem. Pharm. Bull. 2019, 67, 729–732. [Google Scholar] [CrossRef] [Green Version]
- Karimi, S.; Ma, S.; Ramig, K.; Greer, E.M.; Szalda, D.J.; Subramaniam, G. Oxidative Ring-Contraction of 3H-1-Benzazepines to Quinoline Derivatives. Tetrahedron Lett. 2015, 56, 6886–6889. [Google Scholar] [CrossRef]
- Maier, W.F.; Roth, W.; Thies, I.; Schleyer, P.V.R. Hydrogenolysis, iv. gas phase decarboxylation of carboxylic acids. Chem. Ber. 1982, 115, 808–812. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (μM) | |||
|---|---|---|---|---|
| HCT-116 | A375 | A549 | Huh-7 | |
| HO (1) | 0.61 ± 0.03 | 1.34 ± 0.23 | 1.67 ± 0.23 | 1.25 ± 0.08 |
| 2 | >50 | >50 | >50 | >50 |
| 3 | >50 | >50 | >50 | >50 |
| 4 | >50 | >50 | >50 | >50 |
| 5 | >50 | >50 | >50 | >50 |
| 6 | 0.86 ± 0.05 | 4.85 ± 0.42 | 5.47 ± 0.13 | 1.19 ± 0.08 |
| 7 | >50 | >50 | >50 | >50 |
| 8 | 2.19 ± 0.23 | 3.40 ± 0.22 | 43.40 ± 1.08 | 30.75 ± 0.60 |
| 9 | >50 | >50 | >50 | >50 |
| 10 | 2.29 ± 0.27 | 5.79 ± 1.26 | 8.25 ± 0.20 | 6.34 ± 0.32 |
| 11a | 5.95 ± 0.45 | 30.13 ± 1.58 | 27.49 ± 0.89 | >50 |
| 11b | >50 | >50 | >50 | >50 |
| 11c | 10.98 ± 0.34 | 16.42 ± 1.92 | 23.25 ± 0.67 | >50 |
| 11d | >50 | >50 | >50 | >50 |
| 11e | 5.47 ± 0.27 | 7.08 ± 0.42 | 17.98 ± 4.18 | >50 |
| 11f | 10.77 ± 0.42 | 18.56 ± 2.23 | 25.95 ± 0.68 | >50 |
| 12 | 31.88 ± 0.69 | 40.42 ± 7.58 | 34.72 ± 1.94 | >50 |
| 13 | >50 | >50 | >50 | >50 |
| Cisplatin | 4.48 ± 0.18 | 4.04 ± 0.23 | 8.53 ± 0.98 | 15.04 ± 0.15 |
| Compound | IC50 (μM) | SI a | |
|---|---|---|---|
| Huh-7 | L-02 | ||
| 6 | 1.19 ± 0.08 | 67.2 ± 0.43 | 56.5 |
| 10 | 6.34 ± 0.32 | 22.33 ± 1.11 | 3.5 |
| HO (1) | 1.25 ± 0.08 | 3.46 ± 0.36 | 2.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Gong, L.; Chen, C.; Tao, Y.; Zhou, W.; Kong, L.; Luo, J. Semi-Synthesis of Harringtonolide Derivatives and Their Antiproliferative Activity. Molecules 2021, 26, 1380. https://doi.org/10.3390/molecules26051380
Wu X, Gong L, Chen C, Tao Y, Zhou W, Kong L, Luo J. Semi-Synthesis of Harringtonolide Derivatives and Their Antiproliferative Activity. Molecules. 2021; 26(5):1380. https://doi.org/10.3390/molecules26051380
Chicago/Turabian StyleWu, Xiutao, Lijie Gong, Chen Chen, Ye Tao, Wuxi Zhou, Lingyi Kong, and Jianguang Luo. 2021. "Semi-Synthesis of Harringtonolide Derivatives and Their Antiproliferative Activity" Molecules 26, no. 5: 1380. https://doi.org/10.3390/molecules26051380
APA StyleWu, X., Gong, L., Chen, C., Tao, Y., Zhou, W., Kong, L., & Luo, J. (2021). Semi-Synthesis of Harringtonolide Derivatives and Their Antiproliferative Activity. Molecules, 26(5), 1380. https://doi.org/10.3390/molecules26051380