
Novel Donepezil–Arylsulfonamide Hybrids as Multitarget-Directed Ligands for Potential Treatment of Alzheimer’s Disease

 , , and
, , and
Abstract
:
1. Introduction
2. Results and Discussion
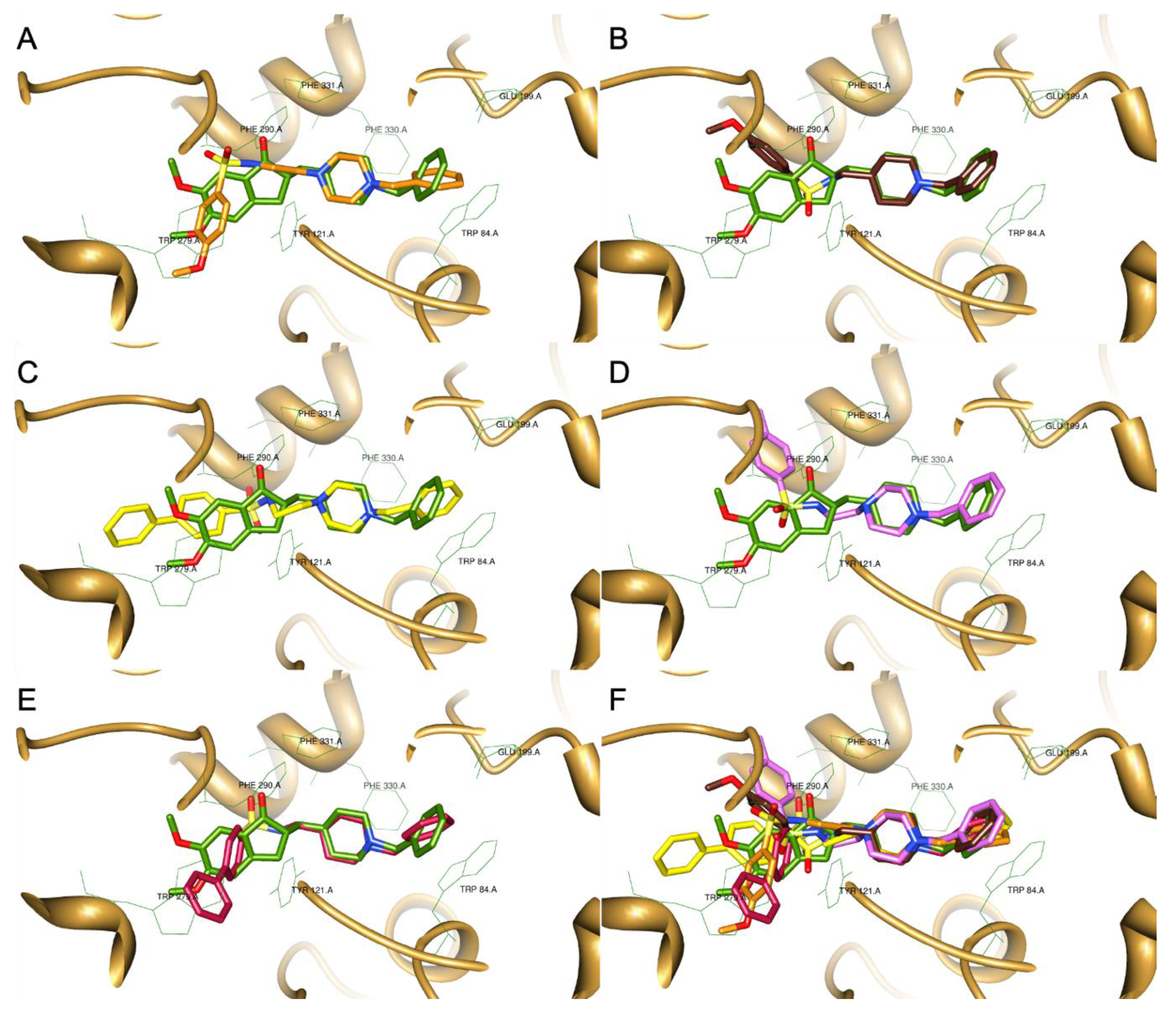
2.1. Molecular Design and Docking
Pharmacokinetic Properties
2.2. Synthesis
2.3. Biological Assays in Solution
2.3.1. Acetylcholinesterase Inhibition
2.3.2. Inhibition of Aβ1–42 Aggregation
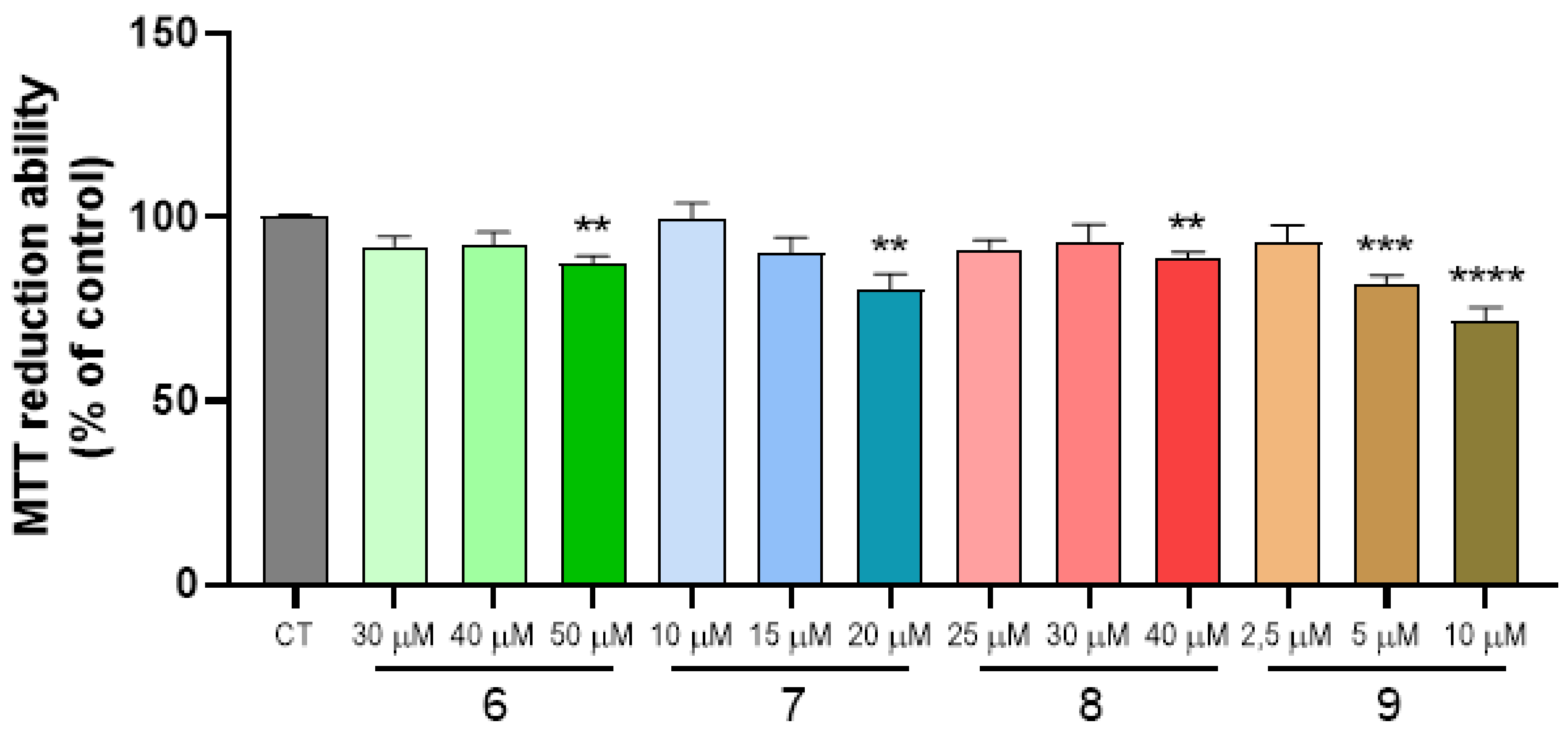
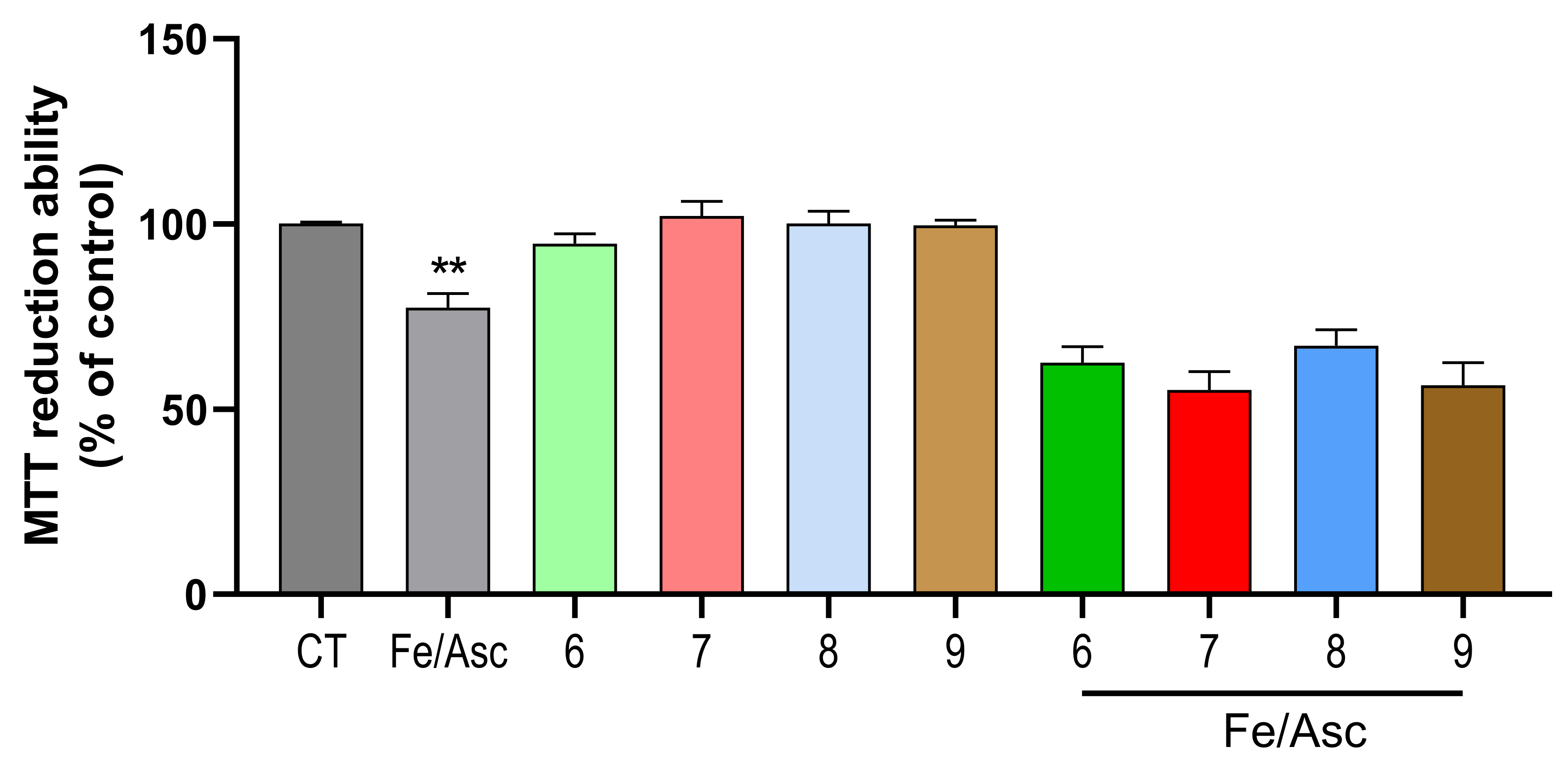
2.4. Cell Viability and Neuroprotection
3. Materials and Methods
3.1. Chemicals and Apparatus
3.2. Molecular Modeling: Docking and Pharmacokinetics Studies
3.3. Synthesis
3.3.1. Synthesis of the Compounds 1 and 2
3.3.2. Synthesis of the Compounds 3 and 4
3.3.3. Synthesis of the Final Conjugates (5–9)
3.4. Biological Assays in Solution
3.4.1. Acetylcholinesterase Inhibition
3.4.2. Inhibition of Aβ1–42 Aggregation
3.5. Cell Viability and Neuroprotection
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cummings, J.L.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline. Alzheimer’s Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Zemek, F.; Drtinova, F.L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar]
- Tan, C.C.; Yu, J.T.; Wang, H.F.; Tan, M.S.; Meng, X.F.; Wang, C.; Jiang, T.; Zhu, X.C.; Tan, L. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: A systematic review and meta-analysis. J. Alzheimer’s Dis. 2014, 41, 615–631. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Fox, N. Defining disease modifying therapy for Alzheimer’s disease. Prev. Alzheimers Dis. 2017, 4, 109–115. [Google Scholar]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments in Alzheimer disease: An update. J. Centr. Neurv. Syst. Dis. 2020, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, L.; Cookson, M.R.; Leonard Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Korabecny, J.; Soukup, O. A perspective on multi-target drugs for Alzheimer’s disease. Trends Pharm. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis in the last decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Wang, N.; Qiu, P.; Cui, W.; Yan, X.; Zhang, B.; He, S. Recent advances in multi-target anti-Alzheimer disease compounds (2013 up to the present). Curr. Med. Chem. 2019, 26, 5684–5710. [Google Scholar] [CrossRef]
- Santos, M.A.; Chand, K.; Chaves, S. Recent progress in repositioning Alzheimer’s disease drugs based on a multitarget strategy. Fut. Med. Chem. 2016, 8, 2113–2142. [Google Scholar] [CrossRef]
- Ismaili, L.; Refouvelet, B.; Benchekroun, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Campiani, G.; Filipic, S.; Agbaba, D.; Esteban, G.; et al. Multitarget compounds bearing tacrine- and donepezil-like structural and functional motifs for the potential treatment of Alzheimer’s disease. Prog. Neurobiol. 2017, 151, 4–34. [Google Scholar] [CrossRef]
- Piemontese, L. New approaches for prevention and treatment of Alzheimer’s disease: A fascinating challenge. Neural Regen. Res. 2017, 12, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Chaves, S.; Várnagy, K.; Santos, M.A. Recent multi-target approaches on the development of anti-Alzheimer’s agents with metal chelation activity. Curr. Med. Chem 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L.; Tomas, D.; Hiremathad, A.; Capriati, V.; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J. Enz. Inhib. Med. Chem. 2018, 33, 1212–1224. [Google Scholar] [CrossRef] [Green Version]
- Chand, K.; Rajeshwari; Candeias, E.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Tacrine–deferiprone hybrids as multi-targetdirected metal chelators against Alzheimer’s disease: A two-in-one drug. Metallomics 2018, 10, 1460–1475. [Google Scholar] [CrossRef]
- Chaves, S.; Resta, S.; Rinaldo, F.; Costa, M.; Josselin, R.; Gwizdala, K.; Piemontese, L.; Capriati, V.; Pereira-Santos, A.R.; Cardoso, S.M.; et al. Design, synthesis and in vitro evaluation of hydroxybenzimidazole-donepezil analogues as multitarget-directed ligands for the treatment of Alzheimer’s disease. Molecules 2020, 25, 985. [Google Scholar] [CrossRef] [Green Version]
- Apaydin, S.; Torok, M. Sulfonamide derivatives as multi-target agents for complex diseases. Bioorg. Med. Chem. Lett. 2019, 29, 2042–2050. [Google Scholar] [CrossRef] [PubMed]
- Bag, S.; Tulsan, R.; Sood, A.; Cho, H.; Redjeb, H.; Zhou, W.; LeVine, H., III; Torok, B.; Torok, M. Sulfonamides as multifunctional agents for Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2015, 25, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Ulus, R.; Kurt, B.Z.; Gazioglu, I.; Kaya, M. Microwave assisted synthesis of novel hybrid tacrine-sulfonamide derivatives and investigation of their antioxidant and anticholinesterase activities. Bioorg. Chem. 2017, 70, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Makhaeva, G.F.; Kovaleva, N.V.; Boltneva, N.P.; Lushchekina, S.V.; Astakhova, T.Y.; Rudakova, E.V.; Proshin, A.N.; Serkov, I.V.; Radchenko, E.V.; Palyulin, V.A.; et al. New hybrids of 4-amino-2,3-polymethylene-quinoline and p-tolylsulfonamide as dual inhibitors of acetyl- and butyryl-cholinesterase and potential multifunctional agents for Alzheimer’s disease treatment. Molecules 2020, 25, 3915. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Rakesh, K.P.; Ravidar, L.; Fang, W.Y.; Qin, H.L. Pharmaceutical and medicinal significance of sulfur (SVI)-containing motifs for drug discovery: A critical review. Eur. J. Med. Chem. 2019, 162, 679–734. [Google Scholar] [CrossRef] [PubMed]
- QikProp, Version 2.5; Schrödinger, LLC: New York, NY, USA, 2005.
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Zagidullin, R.N. Reactions of N-(Β-aminoethyl)piperazine and its derivatives. Chem. Heterocycl. Compd. 1991, 27, 309–312. [Google Scholar] [CrossRef]
- Piemontese, L.; Sergio, R.; Rinaldo, F.; Brunetti, L.; Perna, F.M.; Santos, M.A.; Capriati, V. Deep eutectic solvents as effective reaction media for the synthesis of 2-hydroxyphenylbenzimidazole-based scaffolds en route to donepezil-like compounds. Molecules 2020, 25, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, S.; Bartolini, M.; Ceccarini, L.; Piazzi, L.; Gobbi, S.; Cavalli, A.; Recanatini, M.; Andrisano, V.; Rampa, A. Targeting Alzheimer’s disease: Novel indanone hybrids bearing a pharmacophoric fragment of AP2238. Bioorg. Med. Chem. 2010, 18, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [Green Version]
- Peña-Bautista, C.; Tirle, T.; López-Nogueroles, M.; Vento, M.; Baquero, M.; Cháfer-Pericás, C. Oxidative damage of DNA as early marker of Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 6136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armarego, W.L.F.; Perrin, D.D. Purification of Laboratory Chemicals, 4th ed.; Butterworth-Heinemann: Oxford, UK, 1999. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept (R)): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Maestro, Version 9.3; Schrödinger Inc.: Portland, OR, USA, 2012.
- Hassinen, T.; Perakyla, M. New energy terms for reduced protein models implemented in an off-lattice force field. J. Comp. Chem. 2001, 22, 1229–1242. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comp. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Quintanova, C.; Keri, R.S.; Marques, S.M.; G-Fernandes, M.; Cardoso, S.M.; Serralheiro, M.L.; Santos, M.A. Design, synthesis and bioevaluation of tacrine hybrids with cinnamate and cinnamylidene acetate derivatives as potential anti-Alzheimer drugs. MedChemComm 2015, 6, 1969–1977. [Google Scholar] [CrossRef]
- Sebestik, J.; Marques, S.M.; Fale, P.L.; Santos, S.; Arduino, D.M.; Cardoso, S.M.; Oliveira, C.R.; Serralheiro, M.L.M.; Santos, M.A. Bifunctional phenolic-choline conjugates as anti-oxidants and acetylcholinesterase inhibitors. J. Enz. Inhib. Med. Chem. 2011, 26, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Chand, K.; Esteves, A.R.; Cardoso, S.M.; Ramsay, R.R.; Chaves, S.; Keri, R.S.; Santos, M.A. Tacrine-allyl/propargylcysteine-benzothiazole trihybrids as potential anti-Alzheimer’s drug candidates. RSC Adv. 2016, 6, 53519–53532. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MW(Da) | clog P | log BB | Caco Permeability (nm/s) | CNS |
|---|---|---|---|---|---|
| 5 | 373.512 | 2.259 | 0.123 | 114 | + |
| 6 | 389.512 | 1.972 | 0.094 | 116 | + |
| 7 | 435.583 | 3.575 | 0.021 | 118 | + |
| 8 | 374.497 | 3.080 | −0.354 | 363 | + |
| 9 | 420.568 | 4.686 | −0.319 | 457 | + |
| DNP | 379.50 | 4.269 | 0.132 | 893 | + |
 | X | R | n | AChE Inhib.a IC50 (μM) ± SD | Aβ1–42 Aggreg. Inhib.b,c (%) |
|---|---|---|---|---|---|
| 5 | N | Me | 2 | ≥100 | 41.8 |
| 6 | N | OMe | 2 | 89 ± 9 | 37.2 |
| 7 | N | Ph | 2 | 38.7 ± 0.2 | 34.6 |
| 8 | CH | OMe | 1 | 6.2 ± 0.9 | 64.2 |
| 9 | CH | Ph | 1 | 1.6 ± 0.2 | 60.7 |
| Tacrine | − | − | − | 0.15 ± 0.02 | 54.5 |
| DNP d | − | − | − | 0.026 | <5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Queda, F.; Calò, S.; Gwizdala, K.; Magalhães, J.D.; Cardoso, S.M.; Chaves, S.; Piemontese, L.; Santos, M.A. Novel Donepezil–Arylsulfonamide Hybrids as Multitarget-Directed Ligands for Potential Treatment of Alzheimer’s Disease. Molecules 2021, 26, 1658. https://doi.org/10.3390/molecules26061658
Queda F, Calò S, Gwizdala K, Magalhães JD, Cardoso SM, Chaves S, Piemontese L, Santos MA. Novel Donepezil–Arylsulfonamide Hybrids as Multitarget-Directed Ligands for Potential Treatment of Alzheimer’s Disease. Molecules. 2021; 26(6):1658. https://doi.org/10.3390/molecules26061658
Chicago/Turabian StyleQueda, Fausto, Sonia Calò, Karolina Gwizdala, João D. Magalhães, Sandra M. Cardoso, Sílvia Chaves, Luca Piemontese, and M. Amélia Santos. 2021. "Novel Donepezil–Arylsulfonamide Hybrids as Multitarget-Directed Ligands for Potential Treatment of Alzheimer’s Disease" Molecules 26, no. 6: 1658. https://doi.org/10.3390/molecules26061658
APA StyleQueda, F., Calò, S., Gwizdala, K., Magalhães, J. D., Cardoso, S. M., Chaves, S., Piemontese, L., & Santos, M. A. (2021). Novel Donepezil–Arylsulfonamide Hybrids as Multitarget-Directed Ligands for Potential Treatment of Alzheimer’s Disease. Molecules, 26(6), 1658. https://doi.org/10.3390/molecules26061658