
Superoxide Dismutase Administration: A Review of Proposed Human Uses



Abstract
:
1. Introduction
2. Mechanism of SOD Induction and Inactivation
3. The Role of SOD: What We Have Learned from Knock-Out (KO) Mice
4. SOD as a Detoxification Strategy
5. SOD as a Pharmacological Agent
5.1. Ocular Diseases
5.2. Gastrointestinal Diseases
5.3. Renal Diseases
5.4. Metabolic Diseases
5.5. Cardiovascular Diseases
5.6. Respiratory Diseases
5.7. Neurological Diseases
5.8. Skin Diseases
6. SOD Sources
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [CrossRef]
- Landis, G.N.; Tower, J. Superoxide dismutase evolution and life span regulation. Mech. Ageing Dev. 2005, 126, 365–379. [Google Scholar] [CrossRef]
- Collino, M.; Aragno, M.; Mastrocola, R.; Gallicchio, M.; Rosa, A.C.; Dianzani, C.; Danni, O.; Thiemermann, C.; Fantozzi, R. Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur. J. Pharmacol. 2006, 530, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Collino, M.; Rogazzo, M.; Pini, A.; Benetti, E.; Rosa, A.C.; Chiazza, F.; Fantozzi, R.; Bani, D.; Masini, E. Acute treatment with relaxin protects the kidney against ischaemia/reperfusion injury. J. Cell. Mol. Med. 2013, 17, 1494–1505. [Google Scholar] [CrossRef]
- Singh, N.; Gupta, V.K.; Kumar, A.; Sharma, B. Synergistic Effects of Heavy Metals and Pesticides in Living Systems. Front. Chem. 2017, 5, 70. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Jadot, G.; Vaille, A.; Maldonado, J.; Vanelle, P. Clinical pharmacokinetics and delivery of bovine superoxide dismutase. Clin. Pharmacokinet. 1995, 28, 17–25. [Google Scholar] [CrossRef]
- Nelson, S.K.; Bose, S.K.; McCord, J.M. The toxicity of high-dose superoxide dismutase suggests that superoxide can both initiate and terminate lipid peroxidation in the reperfused heart. Free Radic. Biol. Med. 1994, 16, 195–200. [Google Scholar] [CrossRef]
- Squadrito, G.L.; Pryor, W.A. The nature of reactive species in systems that produce peroxynitrite. Chem. Res. Toxicol. 1998, 11, 718–719. [Google Scholar] [CrossRef]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Kumar, A.; Khushboo, R.; Pandey, R.; Sharma, B. Modulation of Superoxide Dismutase Activity by Mercury, Lead, and Arsenic. Biol. Trace Elem. Res. 2020, 196, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Ighodaro, O.M.; Akinloyeb, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 6. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Wadhwani, A. Antioxidant Enzymes and Human Health. In Antioxidant Enzyme; El-Missiry, M.A., Ed.; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Romao, S. Therapeutic value of oral supplementation with melon superoxide dismutase and wheat gliadin combination. Nutrition 2015, 31, 430–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baret, A.; Jadot, G.; Michelson, A.M. Pharmacokinetic and anti-inflammatory properties in the rat of superoxide dismutases (Cu SODs and Mn SOD) from various species. Biochem. Pharmacol. 1984, 33, 2755–2760. [Google Scholar] [CrossRef]
- De Benito, V.; de Barrio, M.; de Lopez-Saez, M.P.; Ordoqui, E.; Prieto-Garcia, A.; Sainza, T.; Baeza, M.L. Anaphylactic shock caused by impurities in orgotein preparations. Allergol. Immunopathol. 2001, 29, 272–275. [Google Scholar] [CrossRef]
- Vouldoukis, I.; Lacan, D.; Kamate, C.; Coste, P.; Calenda, A.; Mazier, D.; Conti, M.; Dugas, B. Antioxidant and anti-inflammatory properties of a Cucumis melo LC. extract rich in superoxide dismutase activity. J. Ethnopharmacol. 2004, 94, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Carillon, J.; Del Rio, D.; Teissedre, P.L.; Cristol, J.P.; Lacan, D.; Rouanet, J.M. Antioxidant capacity and angiotensin I converting enzyme inhibitory activity of a melon concentrate rich in superoxide dismutase. Food Chem. 2012, 135, 1298–1302. [Google Scholar] [CrossRef] [PubMed]
- Stephenie, S.; Chang, Y.P.; Gnanasekaran, A.; Esa, N.M.; Gnanaraj, C. An insight on superoxide dismutase (SOD) from plants for mammalian. J. Funct. Foods 2020, 68, 103917. [Google Scholar] [CrossRef]
- Vouldoukis, I.; Conti, M.; Krauss, P.; Kamate, C.; Blazquez, S.; Tefit, M.; Mazier, D.; Calenda, A.; Dugas, B. Supplementation with gliadin-combined plant superoxide dismutase extract promotes antioxidant defences and protects against oxidative stress. Phytother. Res. 2004, 18, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.C.; Bruni, N.; Meineri, G.; Corsi, D.; Cavi, N.; Gastaldi, D.; Dosio, F. Strategies to expand the therapeutic potential of superoxide dismutase by exploiting delivery approaches. Int. J. Biol. Macromol. 2020, 168, 846–865. [Google Scholar] [CrossRef]
- Bonetta, R. Potential Therapeutic Applications of MnSODs and SOD-Mimetics. Chemistry 2018, 24, 5032–5041. [Google Scholar] [CrossRef] [Green Version]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue Against Oxidative Injury. Antioxid. Redox Signal. 2018, 29, 1691–1724. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Spasojevic, I. 25 years of development of Mn porphyrins—From mimics of superoxide dismutase enzymes to thiol signaling to clinical trials: The story of our life in the USA. J. Porphyr. Phthalocyanines 2019, 23, 1326–1335. [Google Scholar] [CrossRef] [Green Version]
- Batinic-Haberle, I.; Tome, M.E. Thiol regulation by Mn porphyrins, commonly known as SOD mimics. Redox Biol. 2019, 25, 101139. [Google Scholar] [CrossRef] [PubMed]
- Levanon, D.; Lieman-Hurwitz, J.; Dafni, N.; Wigderson, M.; Sherman, L.; Bernstein, Y.; Laver-Rudich, Z.; Danciger, E.; Stein, O.; Groner, Y. Architecture and anatomy of the chromosomal locus in human chromosome 21 encoding the Cu/Zn superoxide dismutase. EMBO J. 1985, 4, 77–84. [Google Scholar] [CrossRef]
- Wan, X.S.; Devalaraja, M.N.; St Clair, D.K. Molecular structure and organization of the human manganese superoxide dismutase gene. DNA Cell. Biol. 1994, 13, 1127–1136. [Google Scholar] [CrossRef]
- Folz, R.J.; Crapo, J.D. Extracellular superoxide dismutase (SOD3): Tissue-specific expression, genomic characterization, and computer-assisted sequence analysis of the human EC SOD gene. Genomics 1994, 22, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; St Clair, D.K. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milani, P.; Gagliardi, S.; Cova, E.; Cereda, C. SOD1 Transcriptional and Posttranscriptional Regulation and Its Potential Implications in ALS. Neurol. Res. Int. 2011, 2011, 458427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houldsworth, A. A Review of the Role of Mitochondrial Manganese Superoxide Dismutase in Human Disorders, such as, Diabetes. J. Endocrinol. Diabetes 2016, 3, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Kirby, J.; Halligan, E.; Baptista, M.J.; Allen, S.; Heath, P.R.; Holden, H.; Barber, S.C.; Loynes, C.A.; Wood-Allum, C.A.; Lunec, J.; et al. Mutant SOD1 alters the motor neuronal transcriptome: Implications for familial ALS. Brain 2005, 128 Pt 7, 1686–1706. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Rho, H.M. The transcriptional activation of the human copper/zinc superoxide dismutase gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin through two different regulator sites, the antioxidant responsive element and xenobiotic responsive element. Mol. Cell. Biochem. 2002, 240, 47–55. [Google Scholar] [CrossRef]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Qaisiya, M.; Coda Zabetta, C.D.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell. Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ren, X.; Simpkins, J.W. Sequential Upregulation of Superoxide Dismutase 2 and Heme Oxygenase 1 by tert-Butylhydroquinone Protects Mitochondria during Oxidative Stress. Mol. Pharmacol. 2015, 88, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, J.; Wu, S.; Jin, C.; Lu, X.; Hu, X.; Sun, Y.; Gao, X.; Cai, Y. Activation of Nrf2/ARE signaling pathway attenuates lanthanum chloride induced injuries in primary rat astrocytes. Metallomics 2017, 9, 1120–1131. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Carraway, M.S.; Babiker, A.; Suliman, H.B. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 2008, 103, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkseven, S.; Kruger, A.; Mingone, C.J.; Kaminski, P.; Inaba, M.; Rodella, L.F.; Ikehara, S.; Wolin, M.S.; Abraham, N.G. Antioxidant mechanism of heme oxygenase-1 involves an increase in superoxide dismutase and catalase in experimental diabetes. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H701–H707. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, M.J.; Chung, H.S. Fucoidan reduces oxidative stress by regulating the gene expression of HO1 and SOD1 through the Nrf2/ERK signaling pathway in HaCaT cells. Mol. Med. Rep. 2016, 14, 3255–3260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dell’Orco, M.; Milani, P.; Arrigoni, L.; Pansarasa, O.; Sardone, V.; Maffioli, E.; Polveraccio, F.; Bordoni, M.; Diamanti, L.; Ceroni, M.; et al. Hydrogen peroxide-mediated induction of SOD1 gene transcription is independent from Nrf2 in a cellular model of neurodegeneration. Biochim. Biophys. Acta 2016, 1859, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Robledinos-Anton, N.; Fernandez-Gines, R.; Manda, G.; Cuadrado, A. Activators and Inhibitors of NRF2: A Review of Their Potential for Clinical Development. Oxid. Med. Cell. Longev. 2019, 2019, 9372182. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Collaborative Power of Nrf2 and PPARgamma Activators against Metabolic and Drug-Induced Oxidative Injury. Oxid. Med. Cell. Longev 2017, 2017, 1378175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, R.C.; Standiford, T.J. Nrf2 and PPAR{gamma}: PPARtnering against oxidant-induced lung injury. Am. J. Respir. Crit. Care Med. 2010, 182, 134–135. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Salinas, M.; Martin, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J. Neurosci. 2004, 24, 7324–7334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.; Zhai, Y.; Cheng, Q.; Liu, Y.; Gao, X.; Zhang, T.; Wei, Y.; Zhang, F.; Yin, X. The Akt-FoxO3a-manganese superoxide dismutase pathway is involved in the regulation of oxidative stress in diabetic nephropathy. Exp. Physiol. 2013, 98, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, M.O. Extracellular Superoxide Dismutase: Growth Promoter or Tumor Suppressor? Oxid. Med. Cell. Longev. 2016, 2016, 3612589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinas, M.; Diaz, R.; Abraham, N.G.; Ruiz de Galarreta, C.M.; Cuadrado, A. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J. Biol. Chem. 2003, 278, 13898–13904. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Rojo, A.I.; Salinas, M.; Diaz, R.; Gallardo, G.; Alam, J.; De Galarreta, C.M.; Cuadrado, A. Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 2004, 279, 8919–8929. [Google Scholar] [CrossRef] [Green Version]
- Polvani, S.; Tarocchi, M.; Galli, A. PPARgamma and Oxidative Stress: Con(beta) Catenating NRF2 and FOXO. PPAR Res. 2012, 2012, 641087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakaryan, H.; Sarukhanyan, F.; Barkhudaryan, N. Superoxide dismutase (SOD) and nuclear factor kB (NFkB) are involved in the molecular mechanisms of homeostatic activity of hemorphins in response to endotoxin-induced stress is well documented. FEBS J. 2012, 279, 368. [Google Scholar]
- Li, S.; Mao, Y.; Zhou, T.; Luo, C.; Xie, J.; Qi, W.; Yang, Z.; Ma, J.; Gao, G.; Yang, X. Manganese superoxide dismutase mediates anoikis resistance and tumor metastasis in nasopharyngeal carcinoma. Oncotarget 2016, 7, 32408–32420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.M.; Wu, T.C.; Wang, Y.C.; Cheng, Y.W.; Sheu, G.T.; Chen, C.Y.; Lee, H. Activation of NF-kappaB by SOD2 promotes the aggressiveness of lung adenocarcinoma by modulating NKX2-1-mediated IKKbeta expression. Carcinogenesis 2013, 34, 2655–2663. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Gupta Vallur, P.; Phaeton, R.; Mythreye, K.; Hempel, N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants 2017, 6, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, J.; Zhu, J.; Ritzenthaler, J.D.; Zelko, I.N. Epigenetic regulation of EC-SOD expression in aging lung fibroblasts: Role of histone acetylation. Free Radic. Biol. Med. 2017, 112, 212–223. [Google Scholar] [CrossRef]
- Kamiya, T.; Machiura, M.; Makino, J.; Hara, H.; Hozumi, I.; Adachi, T. Epigenetic regulation of extracellular-superoxide dismutase in human monocytes. Free Radic. Biol. Med. 2013, 61, 197–205. [Google Scholar] [CrossRef]
- Banks, C.J.; Andersen, J.L. Mechanisms of SOD1 regulation by post-translational modifications. Redox Biol. 2019, 26, 101270. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.K.; St Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Xu, J.; Ogura, Y.; Monno, I.; Koya, D. Manganese Superoxide Dismutase Dysfunction and the Pathogenesis of Kidney Disease. Front. Physiol. 2020, 11, 755. [Google Scholar] [CrossRef] [PubMed]
- Culotta, V.C.; Yang, M.; O’Halloran, T.V. Activation of superoxide dismutases: Putting the metal to the pedal. Biochim. Biophys. Acta 2006, 1763, 747–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, S.; Ozumi, K.; Kim, H.W.; Nakagawa, O.; McKinney, R.D.; Folz, R.J.; Zelko, I.N.; Ushio-Fukai, M.; Fukai, T. Novel mechanism for regulation of extracellular SOD transcription and activity by copper: Role of antioxidant-1. Free Radic. Biol. Med. 2009, 46, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, T.; Takeuchi, K.; Fukudome, S.; Hara, H.; Adachi, T. Copper chaperone antioxidant-1, Atox-1, is involved in the induction of SOD3 in THP-1 cells. Biometals 2018, 31, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Lutsenko, S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Qiu, S.; Shi, J.; Wang, S.; Wang, M.; Xu, Y.; Nie, Z.; Liu, C.; Liu, C. A new function of copper zinc superoxide dismutase: As a regulatory DNA-binding protein in gene expression in response to intracellular hydrogen peroxide. Nucleic Acids Res. 2019, 47, 5074–5085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, S.; Petrozziello, T.; Ucci, V.; Amente, S.; Santillo, M.; Mondola, P. Cu-Zn superoxide dismutase activates muscarinic acetylcholine M1 receptor pathway in neuroblastoma cells. Mol. Cell. Neurosci. 2013, 52, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Liu, R.; Liu, C.; Chen, Y. Molecular Mechanism of Lead-Induced Superoxide Dismutase Inactivation in Zebrafish Livers. J. Phys. Chem. B 2014, 118, 14820–14826. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.; Demicheli, V.; Duran, R.; Trujillo, M.; Cervenansky, C.; Freeman, B.A.; Radi, R. Inactivation of human Cu,Zn superoxide dismutase by peroxynitrite and formation of histidinyl radical. Free Radic. Biol. Med. 2004, 37, 813–822. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Crow, J.P.; Thompson, J.A. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 1998, 37, 1613–1622. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almomani, R.; Herkert, J.C.; Posafalvi, A.; Post, J.G.; Boven, L.G.; van der Zwaag, P.A.; Willems, P.; van Veen-Hof, I.H.; Verhagen, J.M.A.; Wessels, M.W.; et al. Homozygous damaging SOD2 variant causes lethal neonatal dilated cardiomyopathy. J. Med. Genet. 2020, 57, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Marecki, J.C.; Parajuli, N.; Crow, J.P.; MacMillan-Crow, L.A. The use of the Cre/loxP system to study oxidative stress in tissue-specific manganese superoxide dismutase knockout models. Antioxid. Redox Signal. 2014, 20, 1655–1670. [Google Scholar] [CrossRef] [Green Version]
- Sunagawa, T.; Shimizu, T.; Matsumoto, A.; Tagashira, M.; Kanda, T.; Shirasawa, T.; Nakaya, H. Cardiac electrophysiological alterations in heart/muscle-specific manganese-superoxide dismutase-deficient mice: Prevention by a dietary antioxidant polyphenol. BioMed Res. Int. 2014, 2014, 704291. [Google Scholar] [CrossRef]
- Fidler, T.P.; Rowley, J.W.; Araujo, C.; Boudreau, L.H.; Marti, A.; Souvenir, R.; Dale, K.; Boilard, E.; Weyrich, A.S.; Abel, E.D. Superoxide Dismutase 2 is dispensable for platelet function. Thromb. Haemost. 2017, 117, 1859–1867. [Google Scholar] [CrossRef]
- Misawa, H.; Nakata, K.; Matsuura, J.; Moriwaki, Y.; Kawashima, K.; Shimizu, T.; Shirasawa, T.; Takahashi, R. Conditional knockout of Mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiol. Dis. 2006, 23, 169–177. [Google Scholar] [CrossRef]
- Case, A.J.; Domann, F.E. Manganese superoxide dismutase is dispensable for post-natal development and lactation in the murine mammary gland. Free Radic. Res. 2012, 46, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Salvador, C.; Yang, H.; Huang, T.T.; Epstein, C.J.; Richardson, A. Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Arch. Biochem. Biophys. 1999, 363, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.D.; Van Remmen, H.; Conrad, C.C.; Huang, T.T.; Epstein, C.J.; Richardson, A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J. Biol. Chem. 1998, 273, 28510–28515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, F.J.; Mira, R.G.; Rovegno, M.; Minniti, A.N.; Cerpa, W. Age-related NMDA signaling alterations in SOD2 deficient mice. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 Pt A, 2010–2020. [Google Scholar] [CrossRef]
- Ho, Y.S.; Gargano, M.; Cao, J.; Bronson, R.T.; Heimler, I.; Hutz, R.J. Reduced fertility in female mice lacking copper-zinc superoxide dismutase. J. Biol. Chem. 1998, 273, 7765–7769. [Google Scholar] [CrossRef] [Green Version]
- Elchuri, S.; Oberley, T.D.; Qi, W.; Eisenstein, R.S.; Jackson Roberts, L.; Van Remmen, H.; Epstein, C.J.; Huang, T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 2005, 24, 367–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Shibuya, S.; Ozawa, Y.; Nojiri, H.; Izuo, N.; Yokote, K.; Shimizu, T. Superoxide dismutase 1 loss disturbs intracellular redox signaling, resulting in global age-related pathological changes. BioMed Res. Int. 2014, 2014, 140165. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Wakamatsu, T.H.; Dogru, M.; Ogawa, Y.; Igarashi, A.; Ibrahim, O.M.; Inaba, T.; Shimizu, T.; Noda, S.; Obata, H.; et al. Age-related dysfunction of the lacrimal gland and oxidative stress: Evidence from the Cu,Zn-superoxide dismutase-1 (Sod1) knockout mice. Am. J. Pathol. 2012, 180, 1879–1896. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, E.M.; Marklund, S.L.; Behndig, A. Enhanced age-related cataract in copper-zinc superoxide dismutase null mice. Clin. Exp. Ophthalmol. 2012, 40, 813–820. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Sun, D.; Li, Z.; Wang, L.; Liu, P. Genetic polymorphisms of superoxide dismutases, catalase, and glutathione peroxidase in age-related cataract. Mol. Vis. 2011, 17, 2325–2332. [Google Scholar] [PubMed]
- Chang, D.; Zhang, X.; Rong, S.; Sha, Q.; Liu, P.; Han, T.; Pan, H. Serum antioxidative enzymes levels and oxidative stress products in age-related cataract patients. Oxid. Med. Cell. Longev. 2013, 2013, 587826. [Google Scholar] [CrossRef] [PubMed]
- Wolf, N.; Penn, P.; Pendergrass, W.; Van Remmen, H.; Bartke, A.; Rabinovitch, P.; Martin, G.M. Age-related cataract progression in five mouse models for anti-oxidant protection or hormonal influence. Exp. Eye Res. 2005, 81, 276–285. [Google Scholar] [CrossRef]
- Keithley, E.M.; Canto, C.; Zheng, Q.Y.; Wang, X.; Fischel-Ghodsian, N.; Johnson, K.R. Cu/Zn superoxide dismutase and age-related hearing loss. Hear. Res. 2005, 209, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Tuerdi, A.; Kinoshita, M.; Kamogashira, T.; Fujimoto, C.; Iwasaki, S.; Shimizu, T.; Yamasoba, T. Manganese superoxide dismutase influences the extent of noise-induced hearing loss in mice. Neurosci. Lett. 2017, 642, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.P.; Tangen, C.M.; Walston, J.; Newman, A.B.; Hirsch, C.; Gottdiener, J.; Seeman, T.; Tracy, R.; Kop, W.J.; Burke, G.; et al. Frailty in older adults: Evidence for a phenotype. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, M146–M156. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Bhaskaran, S.; Espinoza, S.; Brooks, S.V.; McArdle, A.; Jackson, M.J.; Van Remmen, H.; Richardson, A. A new mouse model of frailty: The Cu/Zn superoxide dismutase knockout mouse. Geroscience 2017, 39, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Kinugawa, S.; Wang, Z.; Kaminski, P.M.; Wolin, M.S.; Edwards, J.G.; Kaley, G.; Hintze, T.H. Limited exercise capacity in heterozygous manganese superoxide dismutase gene-knockout mice: Roles of superoxide anion and nitric oxide. Circulation 2005, 111, 1480–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deepa, S.S.; Van Remmen, H.; Brooks, S.V.; Faulkner, J.A.; Larkin, L.; McArdle, A.; Jackson, M.J.; Vasilaki, A.; Richardson, A. Accelerated sarcopenia in Cu/Zn superoxide dismutase knockout mice. Free Radic. Biol. Med. 2019, 132, 19–23. [Google Scholar] [CrossRef]
- Sorce, S.; Stocker, R.; Seredenina, T.; Holmdahl, R.; Aguzzi, A.; Chio, A.; Depaulis, A.; Heitz, F.; Olofsson, P.; Olsson, T.; et al. NADPH oxidases as drug targets and biomarkers in neurodegenerative diseases: What is the evidence? Free Radic. Biol. Med. 2017, 112, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef] [PubMed]
- Restagno, G.; Lombardo, F.; Sbaiz, L.; Mari, C.; Gellera, C.; Alimonti, D.; Calvo, A.; Tarenzi, L.; Chio, A. The rare G93D mutation causes a slowly progressing lower motor neuron disease. Amyotroph. Lateral Scler. 2008, 9, 35–39. [Google Scholar] [CrossRef]
- Calvo, A.; Ilardi, A.; Moglia, C.; Canosa, A.; Carrara, G.; Valentini, C.; Ossola, I.; Brunetti, M.; Restagno, G.; Chio, A. An ALS case with a novel D90N-SOD1 heterozygous missense mutation. Amyotroph. Lateral Scler. 2012, 13, 393–395. [Google Scholar] [CrossRef]
- Canosa, A.; De Marco, G.; Lomartire, A.; Rinaudo, M.T.; Di Cunto, F.; Turco, E.; Barberis, M.; Brunetti, M.; Casale, F.; Moglia, C.; et al. A novel p.Ser108LeufsTer15 SOD1 mutation leading to the formation of a premature stop codon in an apparently sporadic ALS patient: Insights into the underlying pathomechanisms. Neurobiol. Aging 2018, 72, 189.e11–189.e17. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H., Jr.; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef]
- Carlsson, L.M.; Jonsson, J.; Edlund, T.; Marklund, S.L. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc. Natl. Acad. Sci. USA 1995, 92, 6264–6268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gongora, M.C.; Lob, H.E.; Landmesser, U.; Guzik, T.J.; Martin, W.D.; Ozumi, K.; Wall, S.M.; Wilson, D.S.; Murthy, N.; Gravanis, M.; et al. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: A potential mechanism underlying adult respiratory distress syndrome. Am. J. Pathol. 2008, 173, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behndig, A. Corneal endothelial integrity in aging mice lacking superoxide dismutase-1 and/or superoxide dismutase-3. Mol. Vis. 2008, 14, 2025–2030. [Google Scholar] [PubMed]
- Behndig, A.; Karlsson, K.; Brannstrom, T.; Sentman, M.L.; Marklund, S.L. Corneal endothelial integrity in mice lacking extracellular superoxide dismutase. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2784–2788. [Google Scholar]
- Fujiwara, T.; Duscher, D.; Rustad, K.C.; Kosaraju, R.; Rodrigues, M.; Whittam, A.J.; Januszyk, M.; Maan, Z.N.; Gurtner, G.C. Extracellular superoxide dismutase deficiency impairs wound healing in advanced age by reducing neovascularization and fibroblast function. Exp. Dermatol. 2016, 25, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentman, M.L.; Brannstrom, T.; Marklund, S.L. EC-SOD and the response to inflammatory reactions and aging in mouse lung. Free Radic. Biol. Med. 2002, 32, 975–981. [Google Scholar] [CrossRef]
- Kwon, M.J.; Jeon, Y.J.; Lee, K.Y.; Kim, T.Y. Superoxide dismutase 3 controls adaptive immune responses and contributes to the inhibition of ovalbumin-induced allergic airway inflammation in mice. Antioxid. Redox Signal. 2012, 17, 1376–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wert, K.J.; Velez, G.; Cross, M.R.; Wagner, B.A.; Teoh-Fitzgerald, M.L.; Buettner, G.R.; McAnany, J.J.; Olivier, A.; Tsang, S.H.; Harper, M.M.; et al. Extracellular superoxide dismutase (SOD3) regulates oxidative stress at the vitreoretinal interface. Free Radic. Biol. Med. 2018, 124, 408–419. [Google Scholar] [CrossRef] [Green Version]
- Tan, R.J.; Zhou, D.; Xiao, L.; Zhou, L.; Li, Y.; Bastacky, S.I.; Oury, T.D.; Liu, Y. Extracellular Superoxide Dismutase Protects against Proteinuric Kidney Disease. J. Am. Soc. Nephrol. 2015, 26, 2447–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.A.; Laskin, D.L.; Smith, C.V.; Robertson, F.M.; Allen, E.M.; Doorn, J.A.; Slikker, W. Nitrative and oxidative stress in toxicology and disease. Toxicol. Sci. 2009, 112, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Andrade, R.J.; Lucena, M.I.; Fernandez, M.C.; Pelaez, G.; Pachkoria, K.; Garcia-Ruiz, E.; Garcia-Munoz, B.; Gonzalez-Grande, R.; Pizarro, A.; Duran, J.A.; et al. Drug-induced liver injury: An analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology 2005, 129, 512–521. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver; Andrade, R.J.; Aithal, G.P.; Björnsson, E.S.; Kaplowitz, N.; Kullak-Ublick, G.A.; Larrey, D.; Karlsen, T.H. EASL Clinical Practice Guidelines: Drug-induced liver injury. J. Hepatol. 2019, 70, 1222–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, H. Emerging novel therapies against paracetamol (acetaminophen) hepatotoxicity. EBioMedicine 2019, 46, 9–10. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, K.; Kumagai, K.; Ito, K.; Arakawa, S.; Ando, Y.; Oda, S.; Yamoto, T.; Manabe, S. Sensitivity of liver injury in heterozygous Sod2 knockout mice treated with troglitazone or acetaminophen. Toxicol. Pathol. 2009, 37, 193–200. [Google Scholar] [CrossRef]
- Agarwal, R.; MacMillan-Crow, L.A.; Rafferty, T.M.; Saba, H.; Roberts, D.W.; Fifer, E.K.; James, L.P.; Hinson, J.A. Acetaminophen-induced hepatotoxicity in mice occurs with inhibition of activity and nitration of mitochondrial manganese superoxide dismutase. J. Pharmacol. Exp. Ther. 2011, 337, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, A.; Lebofsky, M.; Weinman, S.A.; Jaeschke, H. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol. Appl. Pharmacol. 2011, 251, 226–233. [Google Scholar] [CrossRef] [Green Version]
- Trnka, J.; Blaikie, F.H.; Smith, R.A.; Murphy, M.P. A mitochondria-targeted nitroxide is reduced to its hydroxylamine by ubiquinol in mitochondria. Free Radic. Biol. Med. 2008, 44, 1406–1419. [Google Scholar] [CrossRef]
- Ge, Z.; Wang, C.; Zhang, J.; Li, X.; Hu, J. Tempol Protects Against Acetaminophen Induced Acute Hepatotoxicity by Inhibiting Oxidative Stress and Apoptosis. Front. Physiol. 2019, 10, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedda, S.; Laurent, A.; Conti, F.; Chereau, C.; Tran, A.; Tran-Van Nhieu, J.; Jaffray, P.; Soubrane, O.; Goulvestre, C.; Calmus, Y.; et al. Mangafodipir prevents liver injury induced by acetaminophen in the mouse. J. Hepatol. 2003, 39, 765–772. [Google Scholar] [CrossRef]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St Clair, D.; Batinic-Haberle, I. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta 2012, 1822, 794–814. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Farhood, A.; Jaeschke, H. Mitochondria-targeted antioxidant Mito-Tempo protects against acetaminophen hepatotoxicity. Arch. Toxicol. 2017, 91, 761–773. [Google Scholar] [CrossRef]
- Investigators, P.O.P.T.; Dear, J. Randomised open label exploratory, safety and tolerability study with calmangafodipir in patients treated with the 12-h regimen of N-acetylcysteine for paracetamol overdose-the PP100-01 for Overdose of Paracetamol (POP) trial: Study protocol for a randomised controlled trial. Trials 2019, 20, 27. [Google Scholar]
- Jaeschke, H.; Akakpo, J.Y.; Umbaugh, D.S.; Ramachandran, A. Novel Therapeutic Approaches Against Acetaminophen-induced Liver Injury and Acute Liver Failure. Toxicol. Sci. 2020, 174, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasi, A.; Albano, E.; Banni, S.; Botti, B.; Corongiu, F.; Dessi, M.A.; Iannone, A.; Vannini, V.; Dianzani, M.U. Free-radical metabolism of carbon tetrachloride in rat liver mitochondria. A study of the mechanism of activation. Biochem. J. 1987, 246, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Cemek, M.; Aymelek, F.; Buyukokuroglu, M.E.; Karaca, T.; Buyukben, A.; Yilmaz, F. Protective potential of Royal Jelly against carbon tetrachloride induced-toxicity and changes in the serum sialic acid levels. Food Chem. Toxicol. 2010, 48, 2827–2832. [Google Scholar] [CrossRef]
- Poli, G.; Albano, E.; Dianzani, M.U. The role of lipid peroxidation in liver damage. Chem. Phys. Lipids 1987, 45, 117–142. [Google Scholar] [CrossRef]
- Wang, Y.H.; Xu, X.J.; Li, H.L. Hepatoprotective effects of Mimic of Manganese superoxide dismutase against carbon tetrachloride-induced hepatic injury. Int. Immunopharmacol. 2014, 22, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Degoul, F.; Sutton, A.; Mansouri, A.; Cepanec, C.; Degott, C.; Fromenty, B.; Beaugrand, M.; Valla, D.; Pessayre, D. Homozygosity for alanine in the mitochondrial targeting sequence of superoxide dismutase and risk for severe alcoholic liver disease. Gastroenterology 2001, 120, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Wang, L.Y.; Chang, C.H.; Perng, C.L.; Lin, H.C. Superoxide Dismutase 2 Genetic Variation as a Susceptibility Risk Factor for Alcoholic Cirrhosis. Alcohol Alcohol. 2016, 51, 633–637. [Google Scholar] [CrossRef]
- Kessova, I.G.; Ho, Y.S.; Thung, S.; Cederbaum, A.I. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology 2003, 38, 1136–1145. [Google Scholar] [CrossRef]
- Wheeler, M.D.; Kono, H.; Yin, M.; Rusyn, I.; Froh, M.; Connor, H.D.; Mason, R.P.; Samulski, R.J.; Thurman, R.G. Delivery of the Cu/Zn-superoxide dismutase gene with adenovirus reduces early alcohol-induced liver injury in rats. Gastroenterology 2001, 120, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, G.; Perriotte-Olson, C.; Casey, C.A.; Donohue, T.M., Jr.; Talmon, G.A.; Harris, E.N.; Kabanov, A.V.; Saraswathi, V. Effect of nanoformulated copper/zinc superoxide dismutase on chronic ethanol-induced alterations in liver and adipose tissue. Alcohol 2019, 79, 71–79. [Google Scholar] [CrossRef]
- Le Quéré, S.; Lacan, D.; Lemaire, B.; Carillon, J.; Schmitt, K. The role of superoxide dismutase (SOD) in skin disorders. Nutrafoods 2014, 13, 13–27. [Google Scholar] [CrossRef]
- Sasaki, H.; Akamatsu, H.; Horio, T. Protective role of copper, zinc superoxide dismutase against UVB-induced injury of the human keratinocyte cell line HaCaT. J. Investig. Dermatol. 2000, 114, 502–507. [Google Scholar] [CrossRef]
- Takahashi, H.; Hashimoto, Y.; Aoki, N.; Kinouchi, M.; Ishida-Yamamoto, A.; Iizuka, H. Copper, zinc-superoxide dismutase protects from ultraviolet B-induced apoptosis of SV40-transformed human keratinocytes: The protection is associated with the increased levels of antioxidant enzymes. J. Dermatol. Sci. 2000, 23, 12–21. [Google Scholar] [CrossRef]
- Oh, C.T.; Lee, D.; Koo, K.; Lee, J.; Yoon, H.S.; Choi, Y.M.; Kwon, T.R.; Kim, B.J. Superoxide dismutase 1 inhibits alpha-melanocyte stimulating hormone and ultraviolet B-induced melanogenesis in murine skin. Ann. Dermatol. 2014, 26, 681–687. [Google Scholar] [CrossRef] [Green Version]
- Shofian, N.M.; Hamid, A.A.; Osman, A.; Saari, N.; Anwar, F.; Pak Dek, M.S.; Hairuddin, M.R. Effect of freezedrying on the antioxidant compounds and antioxidant activity of selected tropical fruits. Int. J. Mol. Sci. 2011, 12, 4678–4692. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, S.; Rao, P.; Bradshaw, J.; Weller, R. Topical application of superoxide dismutase mediated by HIV-TAT peptide attenuates UVB-induced damages in human skin. Eur. J. Pharm. Biopharm. 2016, 107, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Mehta, G.; Vasiliou, V. Antioxidant defenses in the ocular surface. Ocul. Surf. 2009, 7, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Grumetto, L.; Del Prete, A.; Ortosecco, G.; Barbato, F.; Del Prete, S.; Borrelli, A.; Schiattarella, A.; Mancini, R.; Mancini, A. Study on the Protective Effect of a New Manganese Superoxide Dismutase on the Microvilli of Rabbit Eyes Exposed to UV Radiation. BioMed Res. Int. 2015, 2015, 973197. [Google Scholar] [CrossRef] [Green Version]
- Tasli, N.G.; Cimen, F.K.; Karakurt, Y.; Ucak, T.; Mammadov, R.; Suleyman, B.; Kurt, N.; Suleyman, H. Protective effects of Rutin against methanol induced acute toxic optic neuropathy: An experimental study. Int. J. Ophthalmol. 2018, 11, 780–785. [Google Scholar] [PubMed]
- Setiohadji, B.; Irfani, I.; Rifada, M.; Virgana, R.; Kartasasmita, A.S. The Superoxide Dismutase Mimetic TEMPOL and Its Effect on Retinal Ganglion Cells in Experimental Methanol-Intoxicated Rats. Ophthalmol. Ther. 2018, 7, 167–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treichel, J.L.; Henry, M.M.; Skumatz, C.M.; Eells, J.T.; Burke, J.M. Antioxidants and ocular cell type differences in cytoprotection from formic acid toxicity in vitro. Toxicol. Sci. 2004, 82, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Murnane, J.P. Role of induced genetic instability in the mutagenic effects of chemicals and radiation. Mutat. Res. 1996, 367, 11–23. [Google Scholar] [CrossRef]
- Emerit, J.; Michelson, A.M.; Robert, H.G.; Chomette, G.; Guerin, R.A.; Blondon, J.; Bertrand, M. Superoxide dismutase treatment of 2 cases of radiation-induced sclerosis. Sem. Hop. 1983, 59, 277–281. [Google Scholar]
- Pajović, B.; Snežana, P.; Jelena, K.; Radojčić, B.; Borojević, D.; Radošević-Jelić, M. Role of superoxide dismutase in individualization of breast cancer radiation therapy protocols. Arch. Oncol. 2003, 11, 2. [Google Scholar] [CrossRef] [Green Version]
- Joksic, G.; Pajovic, S.B.; Stankovic, M.; Pejic, S.; Kasapovic, J.; Cuttone, G.; Calonghi, N.; Masotti, L.; Kanazir, D.T. Chromosome aberrations, micronuclei, and activity of superoxide dismutases in human lymphocytes after irradiation in vitro. Cell. Mol. Life Sci. 2000, 57, 842–850. [Google Scholar] [CrossRef]
- Leu, D.; Spasojevic, I.; Nguyen, H.; Deng, B.; Tovmasyan, A.; Weitner, T.; Sampaio, R.S.; Batinic-Haberle, I.; Huang, T.T. CNS bioavailability and radiation protection of normal hippocampal neurogenesis by a lipophilic Mn porphyrin-based superoxide dismutase mimic, MnTnBuOE-2-PyP(5). Redox Biol. 2017, 12, 864–871. [Google Scholar] [CrossRef]
- Tovmasyan, A.; Sheng, H.; Weitner, T.; Arulpragasam, A.; Lu, M.; Warner, D.S.; Vujaskovic, Z.; Spasojevic, I.; Batinic-Haberle, I. Design, mechanism of action, bioavailability and therapeutic effects of mn porphyrin-based redox modulators. Med. Princ. Pract. 2013, 22, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Cline, J.M.; Dugan, G.; Bourland, J.D.; Perry, D.L.; Stitzel, J.D.; Weaver, A.A.; Jiang, C.; Tovmasyan, A.; Owzar, K.; Spasojevic, I.; et al. Post-Irradiation Treatment with a Superoxide Dismutase Mimic, MnTnHex-2-PyP(5+), Mitigates Radiation Injury in the Lungs of Non-Human Primates after Whole-Thorax Exposure to Ionizing Radiation. Antioxidants 2018, 7, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacVittie, T.J.; Gibbs, A.; Farese, A.M.; Barrow, K.; Bennett, A.; Taylor-Howell, C.; Kazi, A.; Prado, K.; Parker, G.; Jackson, W. III, AEOL 10150 Mitigates Radiation-Induced Lung Injury in the Nonhuman Primate: Morbidity and Mortality are Administration Schedule-Dependent. Radiat. Res. 2017, 187, 298–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Hankey, K.G.; Zhang, P.; Bolduc, D.L.; Bunger, R.; Xiao, M.; Farese, A.M.; MacVittie, T.J. Identifying Circulating and Lung Tissue Cytokines Associated with Thoracic Irradiation and AEOL 10150 Treatment in a Nonhuman Primate Model. Radiat. Res. 2020, 194, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.R.; Zhou, W.X.; Zhang, Y.X. Improvements in SOD mimic AEOL-10150, a potent broad-spectrum antioxidant. Mil. Med. Res. 2018, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Antonic, V.; Rabbani, Z.N.; Jackson, I.L.; Vujaskovic, Z. Subcutaneous administration of bovine superoxide dismutase protects lungs from radiation-induced lung injury. Free Radic. Res. 2015, 49, 1259–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattman, C.L.; Tan, R.J.; Tobolewski, J.M.; Oury, T.D. Increased sensitivity to asbestos-induced lung injury in mice lacking extracellular superoxide dismutase. Free Radic. Biol. Med. 2006, 40, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnoni, A.; Ressel, L.; Rossi, D.; Poli, A.; Arienti, D.; Lombardi, G.; Parolini, O. Conditioned medium from amniotic mesenchymal tissue cells reduces progression of bleomycin-induced lung fibrosis. Cytotherapy 2012, 14, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Liu, Y.; Han, Q.; Jia, M.; Liao, L.; Qi, M.; Zhao, R.C. Injured microenvironment directly guides the differentiation of engrafted Flk-1(+) mesenchymal stem cell in lung. Exp. Hematol. 2007, 35, 1466–1475. [Google Scholar] [CrossRef]
- Gao, F.; Kinnula, V.L.; Myllarniemi, M.; Oury, T.D. Extracellular superoxide dismutase in pulmonary fibrosis. Antioxid. Redox Signal. 2008, 10, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Zhang, J.; Yang, Z.L.; You, H. Extracellular superoxide dismutase increased the therapeutic potential of human mesenchymal stromal cells in radiation pulmonary fibrosis. Cytotherapy 2017, 19, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Yucel, S.; Sahin, B.; Gural, Z.; Olgac, V.; Aksu, G.; Agaoglu, F.; Saglam, E.; Aslay, I.; Darendeliler, E. Impact of Superoxide Dismutase-Gliadin on Radiation-induced Fibrosis: An Experimental Study. In Vivo 2016, 30, 451–456. [Google Scholar] [PubMed]
- Can Trabulus, D.; Altinsoy, E.; Karacetin, D.; Nazli, M.A.; Kelten Talu, C. Preventive role of superoxide dismutase on radiation-induced periprosthetic capsule development. J. Surg. Res. 2018, 231, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Landeen, K.C.; Spanos, W.C.; Gromer, L. Topical superoxide dismutase in posttreatment fibrosis in patients with head and neck cancer. Head Neck 2018, 40, 1400–1405. [Google Scholar] [CrossRef] [PubMed]
- Rattay, T.; Talbot, C.J. Finding the genetic determinants of adverse reactions to radiotherapy. Clin. Oncol. 2014, 26, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Sonis, S.T.; Lee, C.M.; Adkins, D.; Allen, B.G.; Sun, W.; Agarwala, S.S.; Venigalla, M.L.; Chen, Y.; Zhen, W.; et al. Phase 1b/2a Trial of the Superoxide Dismutase Mimetic GC4419 to Reduce Chemoradiotherapy-Induced Oral Mucositis in Patients With Oral Cavity or Oropharyngeal Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 427–435. [Google Scholar] [CrossRef] [Green Version]
- Song, M.K.; Park, M.Y.; Sung, M.K. 5-Fluorouracil-induced changes of intestinal integrity biomarkers in BALB/c mice. J. Cancer Prev. 2013, 18, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.X.; Li, H.L.; Zhang, Y.T.; Wu, S.Y.; Lu, H.L.; Yu, X.L.; Meng, F.G.; Sun, J.H.; Gong, L.K. A new recombinant MS-superoxide dismutase alleviates 5-fluorouracil-induced intestinal mucositis in mice. Acta Pharmacol. Sin. 2020, 41, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Mitazaki, S.; Honma, S.; Suto, M.; Kato, N.; Hiraiwa, K.; Yoshida, M.; Abe, S. Interleukin-6 plays a protective role in development of cisplatin-induced acute renal failure through upregulation of anti-oxidative stress factors. Life Sci. 2011, 88, 1142–1148. [Google Scholar] [CrossRef]
- Yang, Y.I.; Ahn, J.H.; Choi, Y.S.; Choi, J.H. Brown algae phlorotannins enhance the tumoricidal effect of cisplatin and ameliorate cisplatin nephrotoxicity. Gynecol. Oncol. 2015, 136, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hosseinian, S.; Khajavi Rad, A.; Hadjzadeh, M.A.; Mohamadian Roshan, N.; Havakhah, S.; Shafiee, S. The protective effect of Nigella sativa against cisplatin-induced nephrotoxicity in rats. Avicenna J. Phytomed. 2016, 6, 44–54. [Google Scholar]
- Ewees, M.G.; Messiha, B.A.S.; Abdel-Bakky, M.S.; Bayoumi, A.M.A.; Abo-Saif, A.A. Tempol, a superoxide dismutase mimetic agent, reduces cisplatin-induced nephrotoxicity in rats. Drug Chem. Toxicol. 2018, 42, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, S.; Ji, Z.; Xu, H.; Zhao, W.; Xia, Z.; Xu, R. Mechanistic study of mtROS-JNK-SOD2 signaling in bupivacaine-induced neuron oxidative stress. Aging 2020, 12, 13463–13476. [Google Scholar] [CrossRef] [PubMed]
- Carillon, J.; Notin, C.; Schmitt, K.; Simoneau, G.; Lacan, D. Dietary supplementation with a superoxide dismutase-melon concentrate reduces stress, physical and mental fatigue in healthy people: A randomised, double-blind, placebo-controlled trial. Nutrients 2014, 6, 2348–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saby, M.; Gauthier, A.; Barial, S.; Egoumenides, L.; Jover, B. Supplementation with a Bioactive Melon Concentrate in Humans and Animals: Prevention of Oxidative Damages and Fatigue in the Context of a Moderate or Eccentric Physical Activity. Int. J. Environ. Res. Public Health 2020, 17, 1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rencus-Lazar, S.; DeRowe, Y.; Adsi, H.; Gazit, E.; Laor, D. Yeast Models for the Study of Amyloid-Associated Disorders and Development of Future Therapy. Front. Mol. Biosci. 2019, 6, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persichilli, S.; Gervasoni, J.; Di Napoli, A.; Fuso, A.; Nicolia, V.; Giardina, B.; Scarpa, S.; Desiderio, C.; Cavallaro, R.A. Plasma thiols levels in Alzheimer’s disease mice under diet-induced hyperhomocysteinemia: Effect of S-adenosylmethionine and superoxide-dismutase supplementation. J. Alzheimers Dis. 2015, 44, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.G.; Mu, H.J.; Li, Z.; Ma, J.H.; Wang, Y.L. Supression of chronic central pain by superoxide dismutase in rats with spinal cord injury: Inhibition of the NMDA receptor implicated. Exp. Ther. Med. 2014, 8, 1137–1141. [Google Scholar] [CrossRef] [Green Version]
- Kartha, S.; Yan, L.; Weisshaar, C.L.; Ita, M.E.; Shuvaev, V.V.; Muzykantov, V.R.; Tsourkas, A.; Winkelstein, B.A.; Cheng, Z. Superoxide Dismutase-Loaded Porous Polymersomes as Highly Efficient Antioxidants for Treating Neuropathic Pain. Adv. Healthc. Mater. 2017, 6, 1700500. [Google Scholar] [CrossRef]
- Clausen, A.; Doctrow, S.; Baudry, M. Prevention of cognitive deficits and brain oxidative stress with superoxide dismutase/catalase mimetics in aged mice. Neurobiol. Aging 2010, 31, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Clausen, A.; Xu, X.; Bi, X.; Baudry, M. Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer’s disease: Protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J. Alzheimers Dis. 2012, 30, 183–208. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.F.; Guo, F.; Cao, Y.Z.; Shi, W.; Xia, Q. Neuroprotection by manganese superoxide dismutase (MnSOD) mimics: Antioxidant effect and oxidative stress regulation in acute experimental stroke. CNS Neurosci. Ther. 2012, 18, 811–818. [Google Scholar] [CrossRef]
- Dohare, P.; Hyzinski-Garcia, M.C.; Vipani, A.; Bowens, N.H.; Nalwalk, J.W.; Feustel, P.J.; Keller, R.W., Jr.; Jourd’heuil, D.; Mongin, A.A. The neuroprotective properties of the superoxide dismutase mimetic tempol correlate with its ability to reduce pathological glutamate release in a rodent model of stroke. Free Radic. Biol. Med. 2014, 77, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Bernardy, C.C.F.; Zarpelon, A.C.; Pinho-Ribeiro, F.A.; Calixto-Campos, C.; Carvalho, T.T.; Fattori, V.; Borghi, S.M.; Casagrande, R.; Verri, W.A., Jr. Tempol, a Superoxide Dismutase Mimetic Agent, Inhibits Superoxide Anion-Induced Inflammatory Pain in Mice. BioMed Res. Int. 2017, 2017, 9584819. [Google Scholar] [CrossRef] [PubMed]
- Carillon, J.; Rugale, C.; Rouanet, J.M.; Cristol, J.P.; Lacan, D.; Jover, B. Endogenous antioxidant defense induction by melon superoxide dismutase reduces cardiac hypertrophy in spontaneously hypertensive rats. Int. J. Food Sci. Nutr. 2014, 65, 602–609. [Google Scholar] [CrossRef]
- Savalia, K.; Manickam, D.S.; Rosenbaugh, E.G.; Tian, J.; Ahmad, I.M.; Kabanov, A.V.; Zimmerman, M.C. Neuronal uptake of nanoformulated superoxide dismutase and attenuation of angiotensin II-dependent hypertension after central administration. Free Radic. Biol. Med. 2014, 73, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Saraswathi, V.; Ganesan, M.; Perriotte-Olson, C.; Manickam, D.S.; Westwood, R.A.; Zimmerman, M.C.; Ahmad, I.M.; Desouza, C.V.; Kabanov, A.V. Nanoformulated copper/zinc superoxide dismutase attenuates vascular cell activation and aortic inflammation in obesity. Biochem. Biophys. Res. Commun. 2016, 469, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Shin, M.J.; Kim, D.W.; Park, J.; Choi, S.Y.; Kang, Y.H. Blockade of monocyte-endothelial trafficking by transduced Tat-superoxide dismutase protein. Int. J. Mol. Med. 2016, 37, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesh, D.; Kumarathasan, P.; Thomson, E.M.; St-Germain, C.; Blais, E.; Crapo, J.; Vincent, R. Impact of Superoxide Dismutase Mimetic AEOL 10150 on the Endothelin System of Fischer 344 Rats. PLoS ONE 2016, 11, e0151810. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Lu, J.; Liu, J.; Li, J. Local Injections of Superoxide Dismutase Attenuate the Exercise Pressor Reflex in Rats with Femoral Artery Occlusion. Front. Physiol. 2018, 9, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcucci, A.; Ruocco, M.R.; Albano, F.; Granato, G.; Romano, V.; Corso, G.; Bancone, C.; De Vendittis, E.; Della Corte, A.; Montagnani, S. Analysis of extracellular superoxide dismutase and Akt in ascending aortic aneurysm with tricuspid or bicuspid aortic valve. Eur. J. Histochem. 2014, 58, 2383. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Rashid, J.; Nozik-Grayck, E.; McMurtry, I.F.; Stenmark, K.R.; Ahsan, F. Cocktail of Superoxide Dismutase and Fasudil Encapsulated in Targeted Liposomes Slows PAH Progression at a Reduced Dosing Frequency. Mol. Pharm. 2017, 14, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, K.I.; Tamura, F.; Sugizaki, T.; Kawahara, M.; Kuba, K.; Imai, Y.; Mizushima, T. Evaluation of Lecithinized Superoxide Dismutase for the Prevention of Acute Respiratory Distress Syndrome in Animal Models. Am. J. Respir Cell. Mol. Biol. 2017, 56, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Yeh, D.Y.; Tung, S.P.; Fu, Y.H.; Yang, Y.C.; Wang, J.J. Intravenous superoxide dismutase administration reduces contralateral lung injury induced by unilateral lung ischemia and reperfusion in rats through suppression of activity and protein expression of matrix metalloproteases. Transplant. Proc. 2015, 47, 1083–1086. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Liao, F.T.; Cheng, H.M.; Sung, S.H.; Yang, Y.C.; Wang, J.J. Intravenous superoxide dismutase as a protective agent to prevent impairment of lung function induced by high tidal volume ventilation. BMC Pulm. Med. 2017, 17, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantino, L.; Goncalves, R.C.; Giombelli, V.R.; Tomasi, C.D.; Vuolo, F.; Kist, L.W.; de Oliveira, G.M.; Pasquali, M.A.; Bogo, M.R.; Mauad, T.; et al. Regulation of lung oxidative damage by endogenous superoxide dismutase in sepsis. Intensive Care Med. Exp. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himori, K.; Abe, M.; Tatebayashi, D.; Lee, J.; Westerblad, H.; Lanner, J.T.; Yamada, T. Superoxide dismutase/catalase mimetic EUK-134 prevents diaphragm muscle weakness in monocrotalin-induced pulmonary hypertension. PLoS ONE 2017, 12, e0169146. [Google Scholar] [CrossRef]
- Villegas, L.R.; Kluck, D.; Field, C.; Oberley-Deegan, R.E.; Woods, C.; Yeager, M.E.; El Kasmi, K.C.; Savani, R.C.; Bowler, R.P.; Nozik-Grayck, E. Superoxide dismutase mimetic, MnTE-2-PyP, attenuates chronic hypoxia-induced pulmonary hypertension, pulmonary vascular remodeling, and activation of the NALP3 inflammasome. Antioxid. Redox Signal. 2013, 18, 1753–1764. [Google Scholar] [CrossRef] [Green Version]
- Zhao, N.; Feng, Z.; Shao, M.; Cao, J.; Wang, F.; Liu, C. Stability Profiles and Therapeutic Effect of Cu/Zn Superoxide Dismutase Chemically Coupled to O-Quaternary Chitosan Derivatives against Dextran Sodium Sulfate-Induced Colitis. Int. J. Mol. Sci. 2017, 18, 1121. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, T.; Tanaka, K.; Tasaka, Y.; Namba, T.; Suzuki, J.; Ishihara, T.; Okamoto, S.; Hibi, T.; Takenaga, M.; Igarashi, R.; et al. Therapeutic effect of lecithinized superoxide dismutase against colitis. J. Pharmacol. Exp. Ther. 2009, 328, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.E.; Kim, H.D.; Park, S.Y.; Pan, J.G.; Kim, J.H.; Yum, D.Y. Dietary Supplementation With a Bacillus Superoxide Dismutase Protects Against gamma-Radiation-induced Oxidative Stress and Ameliorates Dextran Sulphate Sodium-induced Ulcerative Colitis in Mice. J. Crohns Colitis 2018, 12, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Mathieu, E.; Bernard, A.S.; Delsuc, N.; Quevrain, E.; Gazzah, G.; Lai, B.; Chain, F.; Langella, P.; Bachelet, M.; Masliah, J.; et al. A Cell-Penetrant Manganese Superoxide Dismutase (MnSOD) Mimic Is Able to Complement MnSOD and Exerts an Antiinflammatory Effect on Cellular and Animal Models of Inflammatory Bowel Diseases. Inorg. Chem. 2017, 56, 2545–2555. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.H.; Dong, J.; Zhang, J.X.; Zhai, J.; Ge, B. Effects of mimic of manganese superoxide dismutase on 2,4,6-trinitrobenzene sulfonic acid-induced colitis in rats. Arch. Pharm. Res. 2016, 39, 1296–1306. [Google Scholar] [CrossRef]
- Chiumiento, A.; Lamponi, S.; Barbucci, R.; Dominguez, A.; Perez, Y.; Villalonga, R. Immobilizing Cu,Zn-superoxide dismutase in hydrogels of carboxymethylcellulose improves its stability and wound healing properties. Biochemistry 2006, 71, 1324–1328. [Google Scholar] [CrossRef]
- Luo, J.D.; Wang, Y.Y.; Fu, W.L.; Wu, J.; Chen, A.F. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation 2004, 110, 2484–2493. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.T.; Sah, S.K.; Zouboulis, C.C.; Kim, T.Y. Inhibitory effects of superoxide dismutase 3 on Propionibacterium acnes-induced skin inflammation. Sci. Rep. 2018, 8, 4024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrahari, G.; Sah, S.K.; Nguyen, C.T.; Choi, S.S.; Kim, H.Y.; Kim, T.Y. Superoxide Dismutase 3 Inhibits LL-37/KLK-5-Mediated Skin Inflammation through Modulation of EGFR and Associated Inflammatory Cascades. J. Investig. Dermatol. 2020, 140, 656–665.e8. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.L.; Dong, X.; Lahiri, A.; Sebastin, S.J.; Batinic-Haberle, I.; Pervaiz, S.; Puhaindran, M.E. MnSOD is implicated in accelerated wound healing upon Negative Pressure Wound Therapy (NPWT): A case in point for MnSOD mimetics as adjuvants for wound management. Redox Biol. 2019, 20, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Zhuang, H.; Hao, Y.; Zhang, L.; Yang, Q.; Liu, Y.; Qi, C.; Wang, S. Poly(N-Isopropyl-Acrylamide)/Poly(gamma-Glutamic Acid) Thermo-Sensitive Hydrogels Loaded with Superoxide Dismutase for Wound Dressing Application. Int. J. Nanomed. 2020, 15, 1939–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, S.K.; Park, K.H.; Yun, C.O.; Kang, K.S.; Kim, T.Y. Effects of Human Mesenchymal Stem Cells Transduced with Superoxide Dismutase on Imiquimod-Induced Psoriasis-Like Skin Inflammation in Mice. Antioxid. Redox Signal. 2016, 24, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, S.K.; Agrahari, G.; Nguyen, C.T.; Kim, Y.S.; Kang, K.S.; Kim, T.Y. Enhanced therapeutic effects of human mesenchymal stem cells transduced with superoxide dismutase 3 in a murine atopic dermatitis-like skin inflammation model. Allergy 2018, 73, 2364–2376. [Google Scholar] [CrossRef] [PubMed]
- Shariev, A.; Menounos, S.; Laos, A.J.; Laxman, P.; Lai, D.; Hua, S.; Zinger, A.; McRae, C.R.; Casbolt, L.S.; Combes, V.; et al. Skin protective and regenerative effects of RM191A, a novel superoxide dismutase mimetic. Redox Biol. 2021, 38, 101790. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.W.; Shen, C.J.; Tung, Y.T.; Chen, H.L.; Chen, Y.H.; Chang, W.H.; Cheng, K.C.; Yang, S.H.; Chen, C.M. Extracellular superoxide dismutase ameliorates streptozotocin-induced rat diabetic nephropathy via inhibiting the ROS/ERK1/2 signaling. Life Sci. 2015, 135, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.A.; Lim, J.H.; Kim, M.Y.; Kim, Y.; Park, H.S.; Kim, H.W.; Choi, B.S.; Chang, Y.S.; Kim, H.W.; Kim, T.Y.; et al. Extracellular Superoxide Dismutase Attenuates Renal Oxidative Stress Through the Activation of Adenosine Monophosphate-Activated Protein Kinase in Diabetic Nephropathy. Antioxid. Redox Signal. 2018, 28, 1543–1561. [Google Scholar] [CrossRef]
- Ding, W.; Wang, B.; Zhang, M.; Gu, Y. Tempol, a Superoxide Dismutase-Mimetic Drug, Ameliorates Progression of Renal Disease in CKD Mice. Cell. Physiol. Biochem. 2015, 36, 2170–2182. [Google Scholar] [CrossRef]
- De Blasio, M.J.; Ramalingam, A.; Cao, A.H.; Prakoso, D.; Ye, J.M.; Pickering, R.; Watson, A.M.D.; de Haan, J.B.; Kaye, D.M.; Ritchie, R.H. The superoxide dismutase mimetic tempol blunts diabetes-induced upregulation of NADPH oxidase and endoplasmic reticulum stress in a rat model of diabetic nephropathy. Eur. J. Pharmacol. 2017, 807, 12–20. [Google Scholar] [CrossRef]
- Nunes, D.V.; Costa, C.A.; De Bem, G.F.; Cordeiro, V.S.; Santos, I.B.; Carvalho, L.C.; Jordao, A.K.; Cunha, A.C.; Ferreira, V.F.; Moura, R.S.; et al. Tempol, a superoxide dismutase-mimetic drug, prevents chronic ischemic renal injury in two-kidney, one-clip hypertensive rats. Clin. Exp. Hypertens. 2018, 40, 721–729. [Google Scholar] [CrossRef]
- Cao, P.; Ito, O.; Ito, D.; Rong, R.; Zheng, Y.; Kohzuki, M. Combination of Exercise Training and SOD Mimetic Tempol Enhances Upregulation of Nitric Oxide Synthase in the Kidney of Spontaneously Hypertensive Rats. Int. J. Hypertens. 2020, 2020, 2142740. [Google Scholar] [CrossRef]
- Carillon, J.; Knabe, L.; Montalban, A.; Stevant, M.; Keophiphath, M.; Lacan, D.; Cristol, J.P.; Rouanet, J.M. Curative diet supplementation with a melon superoxide dismutase reduces adipose tissue in obese hamsters by improving insulin sensitivity. Mol. Nutr. Food Res. 2014, 58, 842–850. [Google Scholar] [CrossRef]
- Decorde, K.; Agne, A.; Lacan, D.; Ramos, J.; Fouret, G.; Ventura, E.; Feillet-Coudray, C.; Cristol, J.P.; Rouanet, J.M. Preventive effect of a melon extract rich in superoxide scavenging activity on abdominal and liver fat and adipokine imbalance in high-fat-fed hamsters. J. Agric. Food Chem. 2009, 57, 6461–6467. [Google Scholar] [CrossRef]
- Natarajan, G.; Perriotte-Olson, C.; Bhinderwala, F.; Powers, R.; Desouza, C.V.; Talmon, G.A.; Yuhang, J.; Zimmerman, M.C.; Kabanov, A.V.; Saraswathi, V. Nanoformulated copper/zinc superoxide dismutase exerts differential effects on glucose vs lipid homeostasis depending on the diet composition possibly via altered AMPK signaling. Transl. Res. 2017, 188, 10–26. [Google Scholar] [CrossRef]
- Perriotte-Olson, C.; Adi, N.; Manickam, D.S.; Westwood, R.A.; Desouza, C.V.; Natarajan, G.; Crook, A.; Kabanov, A.V.; Saraswathi, V. Nanoformulated copper/zinc superoxide dismutase reduces adipose inflammation in obesity. Obesity 2016, 24, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, T.; Kumar, N.; Perriotte-Olson, C.; Casey, C.A.; Donohue, T.M., Jr.; Harris, E.N.; Talmon, G.; Kabanov, A.V.; Saraswathi, V. Nanoformulated SOD1 ameliorates the combined NASH and alcohol-associated liver disease partly via regulating CYP2E1 expression in adipose tissue and liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G428–G438. [Google Scholar] [CrossRef]
- Coudriet, G.M.; Delmastro-Greenwood, M.M.; Previte, D.M.; Marre, M.L.; O’Connor, E.C.; Novak, E.A.; Vincent, G.; Mollen, K.P.; Lee, S.; Dong, H.H.; et al. Treatment with a Catalytic Superoxide Dismutase (SOD) Mimetic Improves Liver Steatosis, Insulin Sensitivity, and Inflammation in Obesity-Induced Type 2 Diabetes. Antioxidants 2017, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogru, M.; Kojima, T.; Simsek, C.; Tsubota, K. Potential Role of Oxidative Stress in Ocular Surface Inflammation and Dry Eye Disease. Investig. Ophthalmol. Vis. Sci. 2018, 59, DES163–DES168. [Google Scholar] [CrossRef] [Green Version]
- Kost, O.A.; Beznos, O.V.; Davydova, N.G.; Manickam, D.S.; Nikolskaya, I.I.; Guller, A.E.; Binevski, P.V.; Chesnokova, N.B.; Shekhter, A.B.; Klyachko, N.L.; et al. Superoxide Dismutase 1 Nanozyme for Treatment of Eye Inflammation. Oxid. Med. Cell. Longev. 2015, 2015, 5194239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grumetto, L.; Prete, A.D.; Ortosecco, G.; Borrelli, A.; Prete, S.D.; Mancini, A. A Gel Formulation Containing a New Recombinant Form of Manganese Superoxide Dismutase: A Clinical Experience Based on Compassionate Use-Safety of a Case Report. Case Rep. Ophthalmol. Med. 2016, 2016, 7240209. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janko, C.; Filipovic, M.; Munoz, L.E.; Schorn, C.; Schett, G.; Ivanovic-Burmazovic, I.; Herrmann, M. Redox modulation of HMGB1-related signaling. Antioxid. Redox Signal. 2014, 20, 1075–1085. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Zhang, K.; Zhao, L.; Guo, J.; Hu, X.; Chen, Z. Increased serum HMGB1 is related to oxidative stress in patients with atrial fibrillation. J. Int. Med. Res. 2013, 41, 1796–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsung, A.; Klune, J.R.; Zhang, X.; Jeyabalan, G.; Cao, Z.; Peng, X.; Stolz, D.B.; Geller, D.A.; Rosengart, M.R.; Billiar, T.R. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J. Exp. Med. 2007, 204, 2913–2923. [Google Scholar] [CrossRef]
- Mou, K.; Liu, W.; Miao, Y.; Cao, F.; Li, P. HMGB1 deficiency reduces H2 O2 -induced oxidative damage in human melanocytes via the Nrf2 pathway. J. Cell. Mol. Med. 2018, 22, 6148–6156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Zhou, X.; Hu, X.; Jiang, H. H2O2 evokes injury of cardiomyocytes through upregulating HMGB1. Hell. J. Cardiol. 2014, 55, 101–106. [Google Scholar]
- Cui, W.; Hu, G.; Peng, J.; Mu, L.; Liu, J.; Qiao, L. Quercetin Exerted Protective Effects in a Rat Model of Sepsis via Inhibition of Reactive Oxygen Species (ROS) and Downregulation of High Mobility Group Box 1 (HMGB1) Protein Expression. Med. Sci. Monit. 2019, 25, 5795–5800. [Google Scholar] [CrossRef] [PubMed]
- Hassan, H.M.; Al-Wahaibi, L.H.; Elmorsy, M.A.; Mahran, Y.F. Suppression of Cisplatin-Induced Hepatic Injury in Rats Through Alarmin High-Mobility Group Box-1 Pathway by Ganoderma lucidum: Theoretical and Experimental Study. Drug Des. Dev. Ther. 2020, 14, 2335–2353. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, Z.H.; Liu, Y.; Liu, Y.Y. Effects of midazolam combined with sufentanil on injury and expression of HMGB1 and NF-kappaB in rats with pancreatitis. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 2102–2109. [Google Scholar]
- Mohan, S.; Gupta, D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed. Pharmacother. 2018, 108, 1866–1878. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Zhang, Z.; Zhang, P.; Zheng, C.; Zhou, W.; Cui, W.; Xu, L.; Gao, J. Downregulation of HMGB1 is required for the protective role of Nrf2 in EMT-mediated PF. J. Cell. Physiol. 2019, 234, 8862–8872. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Wang, H.; Wang, L. Protective effects of ghrelin against oxidative stress, inducible nitric oxide synthase and inflammation in a mouse model of myocardial ischemia/reperfusion injury via the HMGB1 and TLR4/NF-kappaB pathway. Mol. Med. Rep. 2016, 14, 2764–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, T.; Yue, Y.; Wang, X.; Li, H.; Yan, S. Luteolin Relieved DSS-Induced Colitis in Mice via HMGB1-TLR-NF-kappaB Signaling Pathway. Inflammation 2020, 44, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; Leon, R.; Lopez, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barancik, M.; Gresova, L.; Bartekova, M.; Dovinova, I. Nrf2 as a key player of redox regulation in cardiovascular diseases. Physiol Res. 2016, 65 (Suppl. 1), S1–S10. [Google Scholar] [CrossRef]
- Cho, H.Y.; Gladwell, W.; Wang, X.; Chorley, B.; Bell, D.; Reddy, S.P.; Kleeberger, S.R. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am. J. Respir Crit. Care Med. 2010, 182, 170–182. [Google Scholar] [CrossRef]
- Dovinova, I.; Kvandova, M.; Balis, P.; Gresova, L.; Majzunova, M.; Horakova, L.; Chan, J.Y.; Barancik, M. The role of Nrf2 and PPARgamma in the improvement of oxidative stress in hypertension and cardiovascular diseases. Physiol. Res. 2020, 69 (Suppl. 4), S541–S553. [Google Scholar] [CrossRef]
- Kvandova, M.; Barancik, M.; Balis, P.; Puzserova, A.; Majzunova, M.; Dovinova, I. The peroxisome proliferator-activated receptor gamma agonist pioglitazone improves nitric oxide availability, renin-angiotensin system and aberrant redox regulation in the kidney of pre-hypertensive rats. J. Physiol. Pharmacol. 2018, 69. [Google Scholar] [CrossRef]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Kaluzhny, Y.; Kinuthia, M.W.; Lapointe, A.M.; Truong, T.; Klausner, M.; Hayden, P. Oxidative stress in corneal injuries of different origin: Utilization of 3D human corneal epithelial tissue model. Exp. Eye Res. 2020, 190, 107867. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Simsek, C.; Kojima, T.; Higa, K.; Kawashima, M.; Dogru, M.; Shimizu, T.; Tsubota, K.; Shimazaki, J. The effects of 3% diquafosol sodium eye drop application on meibomian gland and ocular surface alterations in the Cu, Zn-superoxide dismutase-1 (Sod1) knockout mice. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Seen, S.; Tong, L. Dry eye disease and oxidative stress. Acta Ophthalmol. 2018, 96, e412–e420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susila, N.K.; Mahayani, N.M.; Triningrat, A.A.; Widiana, I.G.; Djelantik, A.A.; Jayanegara, W.G. Blood superoxide dismutase (SOD) level has a negative correlation with dry eye (DE) degree. Bali Med. J. 2017, 6, 390–394. [Google Scholar] [CrossRef]
- Jivabhai Patel, S.; Bany-Mohammed, F.; McNally, L.; Valencia, G.B.; Lazzaro, D.R.; Aranda, J.V.; Beharry, K.D. Exogenous Superoxide Dismutase Mimetic Without Scavenging H2O2 Causes Photoreceptor Damage in a Rat Model for Oxygen-Induced Retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1665–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruidenier, L.; Verspaget, H.W. Review article: Oxidative stress as a pathogenic factor in inflammatory bowel disease--radicals or ridiculous? Aliment. Pharmacol. Ther. 2002, 16, 1997–2015. [Google Scholar] [CrossRef] [PubMed]
- Beltran, B.; Nos, P.; Dasi, F.; Iborra, M.; Bastida, G.; Martinez, M.; O’Connor, J.E.; Saez, G.; Moret, I.; Ponce, J. Mitochondrial dysfunction, persistent oxidative damage, and catalase inhibition in immune cells of naive and treated Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Kruidenier, L.; Kuiper, I.; van Duijn, W.; Marklund, S.L.; van Hogezand, R.A.; Lamers, C.B.; Verspaget, H.W. Differential mucosal expression of three superoxide dismutase isoforms in inflammatory bowel disease. J. Pathol. 2003, 201, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Lan, S. Implications of Antioxidant Systems in Inflammatory Bowel Disease. BioMed Res. Int. 2018, 2018, 1290179. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.A.; Bae, E.A.; Hyun, Y.J.; Kim, D.H. Dextran sulfate sodium and 2,4,6-trinitrobenzene sulfonic acid induce lipid peroxidation by the proliferation of intestinal gram-negative bacteria in mice. J. Inflamm. 2010, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, L.; Oudart, H.; Rene, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [Green Version]
- Muscogiuri, G.; Salmon, A.B.; Aguayo-Mazzucato, C.; Li, M.; Balas, B.; Guardado-Mendoza, R.; Giaccari, A.; Reddick, R.L.; Reyna, S.M.; Weir, G.; et al. Genetic disruption of SOD1 gene causes glucose intolerance and impairs beta-cell function. Diabetes 2013, 62, 4201–4207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keaney, J.F., Jr.; Larson, M.G.; Vasan, R.S.; Wilson, P.W.; Lipinska, I.; Corey, D.; Massaro, J.M.; Sutherland, P.; Vita, J.A.; Benjamin, E.J.; et al. Obesity and systemic oxidative stress: Clinical correlates of oxidative stress in the Framingham Study. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Tse, H.M.; Milton, M.J.; Piganelli, J.D. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: Implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic. Biol. Med. 2004, 36, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Mate, A.; Miguel-Carrasco, J.L.; Monserrat, M.T.; Vazquez, C.M. Systemic antioxidant properties of L-carnitine in two different models of arterial hypertension. J. Physiol. Biochem. 2010, 66, 127–136. [Google Scholar] [CrossRef]
- Zhou, F.; Zhong, W.; Xue, J.; Gu, Z.L.; Xie, M.L. Reduction of rat cardiac hypertrophy by osthol is related to regulation of cardiac oxidative stress and lipid metabolism. Lipids 2012, 47, 987–994. [Google Scholar] [CrossRef]
- Dornas, W.C.; Silva, M.; Tavares, R.; de Lima, W.G.; dos Santos, R.C.; Pedrosa, M.L.; Silva, M.E. Efficacy of the superoxide dismutase mimetic tempol in animal hypertension models: A meta-analysis. J. Hypertens. 2015, 33, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Wassmann, K.; Nickenig, G. Modulation of oxidant and antioxidant enzyme expression and function in vascular cells. Hypertension 2004, 44, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.B. The circumventricular organs and the central actions of angiotensin. Neuroendocrinology 1981, 32, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Davisson, R.L. Redox signaling in central neural regulation of cardiovascular function. Prog. Biophys. Mol. Biol. 2004, 84, 125–149. [Google Scholar] [CrossRef] [PubMed]
- Decharatchakul, N.; Settasatian, C.; Settasatian, N.; Komanasin, N.; Kukongviriyapan, U.; Intharapetch, P.; Senthong, V.; Sawanyawisuth, K. Association of combined genetic variations in SOD3, GPX3, PON1, and GSTT1 with hypertension and severity of coronary artery disease. Heart Vessel. 2020, 35, 918–929. [Google Scholar] [CrossRef]
- Mansego, M.L.; Solar Gde, M.; Alonso, M.P.; Martinez, F.; Saez, G.T.; Escudero, J.C.; Redon, J.; Chaves, F.J. Polymorphisms of antioxidant enzymes, blood pressure and risk of hypertension. J. Hypertens. 2011, 29, 492–500. [Google Scholar] [CrossRef]
- Dong, X.; Li, D.; Liu, H.; Zhao, Y. SOD3 and eNOS genotypes are associated with SOD activity and NOx. Exp. Ther. Med. 2014, 8, 328–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersson, J.; Glenny, R.W. Gas exchange and ventilation-perfusion relationships in the lung. Eur. Respir. J. 2014, 44, 1023–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnula, V.L.; Crapo, J.D. Superoxide dismutases in the lung and human lung diseases. Am. J. Respir. Crit. Care Med. 2003, 167, 1600–1619. [Google Scholar] [CrossRef]
- Montani, D.; Gunther, S.; Dorfmuller, P.; Perros, F.; Girerd, B.; Garcia, G.; Jais, X.; Savale, L.; Artaud-Macari, E.; Price, L.C.; et al. Pulmonary arterial hypertension. Orphanet J. Rare Dis. 2013, 8, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Channick, R.N. Combination therapy in pulmonary arterial hypertension. Am. J. Cardiol. 2013, 111 (Suppl. 8), 16C–20C. [Google Scholar] [CrossRef]
- Georgieva, G.S.; Kurata, S.; Ikeda, S.; Eishi, Y.; Mitaka, C.; Imai, T. Nonischemic lung injury by mediators from unilateral ischemic reperfused lung: Ameliorating effect of tumor necrosis factor-alpha-converting enzyme inhibitor. Shock 2007, 27, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Soccal, P.M.; Gasche, Y.; Miniati, D.N.; Hoyt, G.; Berry, G.J.; Doyle, R.L.; Theodore, J.; Robbins, R.C. Matrix metalloproteinase inhibition decreases ischemia-reperfusion injury after lung transplantation. Am. J. Transplant. 2004, 4, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Savla, U.; Sporn, P.H.; Waters, C.M. Cyclic stretch of airway epithelium inhibits prostanoid synthesis. Am. J. Physiol. 1997, 273, L1013–L1019. [Google Scholar] [CrossRef]
- Salvemini, D.; Cuzzocrea, S. Oxidative stress in septic shock and disseminated intravascular coagulation. Free Radic. Biol. Med. 2002, 33, 1173–1185. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Jiang, Z.G.; Ghanbari, H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013, 14, 24438–24475. [Google Scholar] [CrossRef] [Green Version]
- Ng, F.; Berk, M.; Dean, O.; Bush, A.I. Oxidative stress in psychiatric disorders: Evidence base and therapeutic implications. Int. J. Neuropsychopharmacol. 2008, 11, 851–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, T.P.; Fonseca, F.L.; de Carvalho, M.D.; Godinho, R.M.; de Almeida, F.P.; Saint’Pierre, T.D.; Rey, N.A.; Fernandes, C.; Horn, A., Jr.; Pereira, M.D. Metal-based superoxide dismutase and catalase mimics reduce oxidative stress biomarkers and extend life span of Saccharomyces cerevisiae. Biochem. J. 2017, 474, 301–315. [Google Scholar] [CrossRef]
- Sterniczuk, R.; Antle, M.C.; Laferla, F.M.; Dyck, R.H. Characterization of the 3xTg-AD mouse model of Alzheimer’s disease: Part 2. Behavioral and cognitive changes. Brain Res. 2010, 1348, 149–155. [Google Scholar] [CrossRef]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in Amyotrophic Lateral Sclerosis: “Ambivalent” Behavior Connected to the Disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Elpers, C.; Reunert, J.; McCormick, M.L.; Mohr, J.; Biskup, S.; Schwartz, O.; Rust, S.; Gruneberg, M.; Seelhofer, A.; et al. SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain 2019, 142, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Abati, E.; Bresolin, N.; Comi, G.; Corti, S. Silence superoxide dismutase 1 (SOD1): A promising therapeutic target for amyotrophic lateral sclerosis (ALS). Expert Opin. Ther. Targets 2020, 24, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Engidawork, E.; Lubec, G. Protein expression in Down syndrome brain. Amino Acids 2001, 21, 331–361. [Google Scholar] [CrossRef]
- Netto, C.B.; Siqueira, I.R.; Fochesatto, C.; Portela, L.V.; da Purificacao Tavares, M.; Souza, D.O.; Giugliani, R.; Goncalves, C.A. S100B content and SOD activity in amniotic fluid of pregnancies with Down syndrome. Clin. Biochem. 2004, 37, 134–137. [Google Scholar] [CrossRef]
- Domingues, N.B.; Mariusso, M.R.; Tanaka, M.H.; Scarel-Caminaga, R.M.; Mayer, M.P.A.; Brighenti, F.L.; Zuanon, A.C.C.; Ibuki, F.K.; Nogueira, F.N.; Giro, E.M.A. Reduced salivary flow rate and high levels of oxidative stress in whole saliva of children with Down syndrome. Spec. Care Dent. 2017, 37, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, M.; Lutter, R.; Eldering, E.; Bos, A.P.; van Woensel, J.B. Effect of oxidative stress on respiratory epithelium from children with Down syndrome. Eur. Respir. J. 2013, 42, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Li, T.; Chen, J.; Liu, Y.; Xiong, F.; Yang, J.; Song, C. Plasma antioxidant enzymes and lipoperoxidation status in children with Down syndrome. Clin. Biochem. 2016, 49, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Muchova, J.; Zitnanova, I.; Durackova, Z. Oxidative stress and Down syndrome. Do antioxidants play a role in therapy? Physiol. Res. 2014, 63, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Lott, I.T. Antioxidants in Down syndrome. Biochim. Biophys. Acta 2012, 1822, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda Revilla, N.; Martinez-Cue, C. Antioxidants in Down Syndrome: From Preclinical Studies to Clinical Trials. Antioxidants 2020, 9, 692. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, C.; Naziroglu, M.; Rodriguez, A.B.; Pariente, J.A. Neuropathic Pain: Delving into the Oxidative Origin and the Possible Implication of Transient Receptor Potential Channels. Front. Physiol. 2018, 9, 95. [Google Scholar] [CrossRef]
- Kurahashi, T.; Fujii, J. Roles of Antioxidative Enzymes in Wound Healing. J. Dev. Biol. 2015, 3, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Campanati, A.; Consales, V.; Orciani, M.; Giuliodori, K.; Ganzetti, G.; Bobyr, I.; Sorgentoni, G.; di Primio, R.; Offidani, A. Role of mesenchymal stem cells in the pathogenesis of psoriasis: Current perspectives. Psoriasis 2017, 7, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeinali, F.; Homaei, A.; Kamrani, E. Sources of marine superoxide dismutases: Characteristics and applications. Int. J. Biol. Macromol. 2015, 79, 627–637. [Google Scholar] [CrossRef]
- Sah, S.K.; Agrahari, G.; Kim, T.Y. Insights into superoxide dismutase 3 in regulating biological and functional properties of mesenchymal stem cells. Cell. Biosci. 2020, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.K.; Elumalai, S. Industrial Production of Superoxide Dismutase (SOD): A Mini Review. J. Probiotics Health 2017, 5, 5. [Google Scholar] [CrossRef]
- Wang, W.; Xia, M.X.; Chen, J.; Yuan, R.; Deng, F.N.; Shen, F.F. Gene Expression Characteristics and Regulation Mechanisms of Superoxide Dismutase and Its Physiological Roles in Plants under Stress. Biochemistry 2016, 81, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Carillon, J.; Jover, B.; Cristol, J.P.; Rouanet, J.M.; Richard, S.; Virsolvy, A. Dietary supplementation with a specific melon concentrate reverses vascular dysfunction induced by cafeteria diet. Food Nutr. Res. 2016, 60, 32729. [Google Scholar] [CrossRef]
- Hou, Z.; Zhao, L.; Wang, Y.; Liao, X. Purification and Characterization of Superoxide Dismutases from Sea Buckthorn and Chestnut Rose. J. Food Sci. 2019, 84, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Chohan, M.; Naughton, D.P.; Opara, E.I. Determination of superoxide dismutase mimetic activity in common culinary herbs. Springerplus 2014, 3, 578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, M.M.; Flickinger, A.G.; Riley, D.P.; Weiss, R.H.; Ryan, U.S. Superoxide dismutase mimetics inhibit neutrophil-mediated human aortic endothelial cell injury in vitro. J. Biol. Chem. 1994, 269, 18535–18540. [Google Scholar] [CrossRef]
- Filograna, R.; Godena, V.K.; Sanchez-Martinez, A.; Ferrari, E.; Casella, L.; Beltramini, M.; Bubacco, L.; Whitworth, A.J.; Bisaglia, M. Superoxide Dismutase (SOD)-mimetic M40403 Is Protective in Cell and Fly Models of Paraquat Toxicity: IMPLICATIONS FOR PARKINSON DISEASE. J. Biol. Chem. 2016, 291, 9257–9267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCord, J.M. Superoxide dismutase, lipid peroxidation, and bell-shaped dose response curves. Dose Response 2008, 6, 223–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsson, J.O.; Ignarro, L.J.; Lundstrom, I.; Jynge, P.; Almen, T. Calmangafodipir [Ca4Mn(DPDP)5], mangafodipir (MnDPDP) and MnPLED with special reference to their SOD mimetic and therapeutic properties. Drug Discov. Today 2015, 20, 411–421. [Google Scholar] [CrossRef] [Green Version]
- Muscoli, C.; Cuzzocrea, S.; Riley, D.P.; Zweier, J.L.; Thiemermann, C.; Wang, Z.Q.; Salvemini, D. On the selectivity of superoxide dismutase mimetics and its importance in pharmacological studies. Br. J. Pharmacol. 2003, 140, 445–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batinic-Haberle, I.; Reboucas, J.S.; Spasojevic, I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlichte, S.L.; Romanova, S.; Katsurada, K.; Kosmacek, E.A.; Bronich, T.K.; Patel, K.P.; Oberley-Deegan, R.E.; Zimmerman, M.C. Nanoformulation of the superoxide dismutase mimic, MnTnBuOE-2-PyP(5+), prevents its acute hypotensive response. Redox Biol. 2020, 36, 101610. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.A.; Fish, B.; Hill, R.P.; Huffman, K.D.; Lazarova, Z.; Mahmood, J.; Medhora, M.; Molthen, R.; Moulder, J.E.; Sonis, S.T.; et al. Salen Mn complexes mitigate radiation injury in normal tissues. Anticancer Agents Med. Chem. 2011, 11, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Xu, P.; Huang, M.; Chen, X.; Zeng, S.; Wang, Q.; Chen, J.; Li, K.; Gao, W.; Liu, R.; et al. The heterocyclic compound Tempol inhibits the growth of cancer cells by interfering with glutamine metabolism. Cell Death Dis. 2020, 11, 312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wang, Q.; Zhou, Q.; Wang, R.; Xu, M.; Wang, H.; Wang, L.; Wilcox, C.S.; Liu, R.; Lai, E.Y. Protective Effect of Tempol on Acute Kidney Injury Through PI3K/Akt/Nrf2 Signaling Pathway. Kidney Blood Press. Res. 2016, 41, 129–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, D.A.D.; Correia, T.M.L.; Pereira, R.; da Silva, R.A.A.; Augusto, O.; Queiroz, R.F. Tempol reduces inflammation and oxidative damage in cigarette smoke-exposed mice by decreasing neutrophil infiltration and activating the Nrf2 pathway. Chem. Biol. Interact. 2020, 329, 109210. [Google Scholar] [CrossRef]
- Kelso, G.F.; Maroz, A.; Cocheme, H.M.; Logan, A.; Prime, T.A.; Peskin, A.V.; Winterbourn, C.C.; James, A.M.; Ross, M.F.; Brooker, S.; et al. A mitochondria-targeted macrocyclic Mn(II) superoxide dismutase mimetic. Chem. Biol. 2012, 19, 1237–1246. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, J.O.G.; Jynge, P.; Ignarro, L.J. May Mangafodipir or Other SOD Mimetics Contribute to Better Care in COVID-19 Patients? Antioxidants 2020, 9, 971. [Google Scholar] [CrossRef] [PubMed]


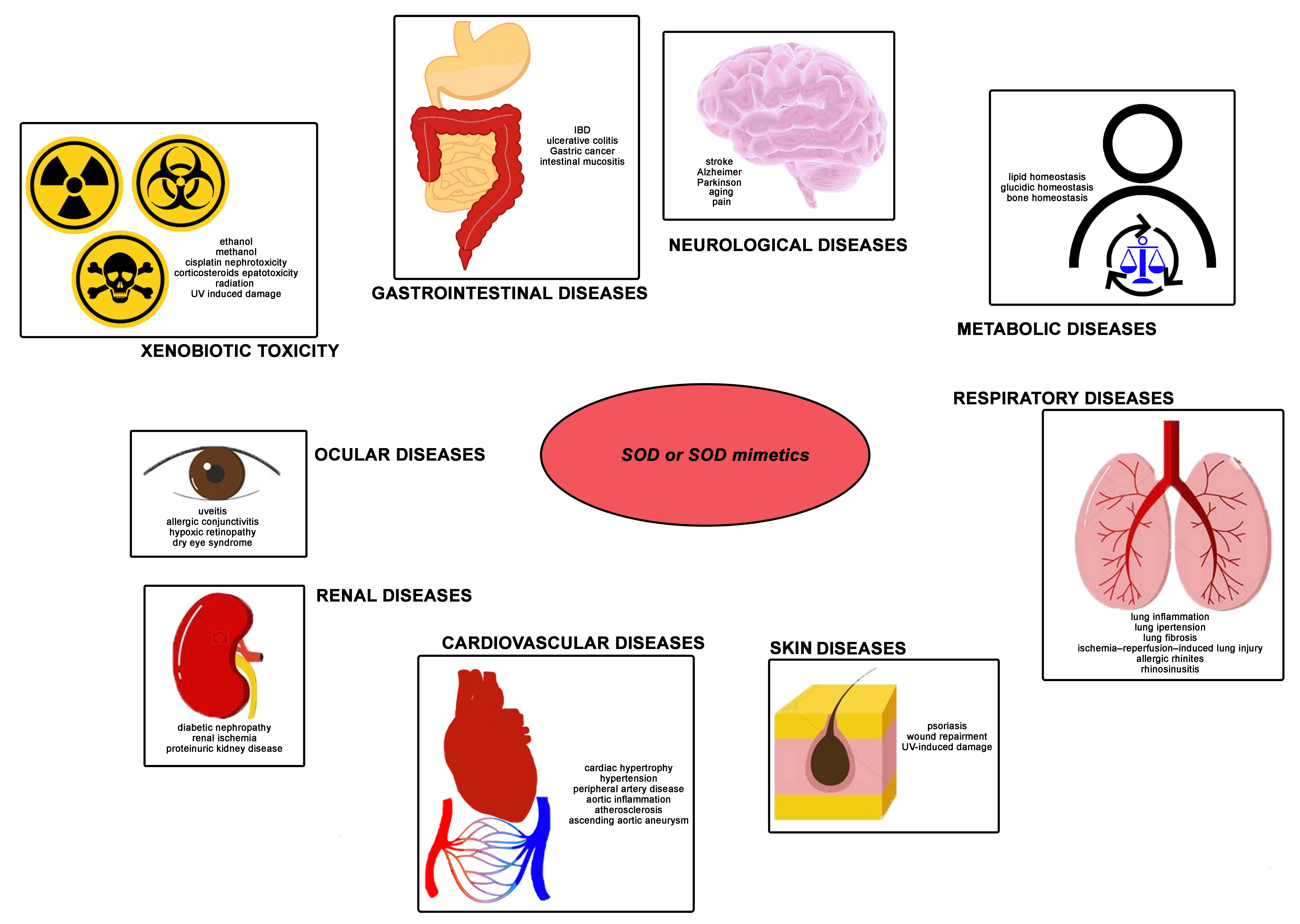{kind=link}
{kind=link}
| Insult | Treatment Tested | Reference(s) |
|---|---|---|
| paracetamol hepatotoxicity | mangafodipir | [125] |
| mito-tempo | [127] | |
| tempol | [124] | |
| calmangafodipir | [128] *, [129] | |
| carbon tetrachloride intoxication | SOD2m | [133] |
| alcohol intoxication | SOD1 | [137] |
| nano-SOD | [138] | |
| methanol intoxication | tempol | [148] |
| UV-induced skin damage | SODB | [139] |
| SOD1 | [140,141,142] | |
| TAT-SOD | [144] | |
| EUK-134 | [143] | |
| UV-induced ocular pathologies | rMnSOD | [146] |
| radiotherapy-induced cytotoxic response | gliadin SOD | [166,167] |
| SOD | [160], [168] * | |
| SOD3 | [165] | |
| GC4419 | [170] * | |
| MnTnBuOE-2-PyP5+ | [154] | |
| MnTDE-2-ImP5+ | [157,158,159] | |
| MnTnHex-2-PyP5+ | [155,156] | |
| SOD3-overexpressing MSCs | [164] | |
| cisplatin-induced oral mucositis | GC4419 | [170] * |
| cisplatin-induced nephrotoxicity | tempol | [175] |
| 5-fluorouracil-induced intestinal mucositis | MS-AOE® | [172] |
| Bupivacaine-induced neurotoxicity | mito-tempo | [177] |
| Application | SOD Formulation | References |
|---|---|---|
| neurological diseases | SODB | [178] *, [179] * |
| SOD1 | [180] | |
| SOD | [181,182] | |
| SOD-loaded porous polymersome | [183] | |
| EUK-207 | [184,185] | |
| MnTM-4-PyP5+ | [186] | |
| tempol | [187,188] | |
| cardiovascular diseases | SODB | [20,189] |
| nano-SOD | [190,191] | |
| TAT-SOD | [192] | |
| MnTDE-2-ImP5+ | [193] | |
| tempol | [194] | |
| SOD3-overexpressing MSCs | [195] | |
| respiratory diseases | CAR-modified liposomes fasudil plus SOD | [196] |
| PC-SOD | [197] | |
| SOD1 | [198,199] | |
| [Fe(HPClNOL)Cl2]NO3 | [200] | |
| EUK-134 | [201] | |
| MnTE-2-PyP5+ | [202] | |
| gastrointestinal diseases | O-HTCC-SOD | [203] |
| PC-SOD | [204] | |
| SOD2 by Bacillus amyloliquefaciens strain | [205] | |
| Mn1 | [206] | |
| SOD2m | [207] | |
| skin diseases | SOD1 | [208] |
| SOD2 | [209] | |
| SOD3 | [210,211] | |
| MnTE-2-PyP5+ | [212] | |
| SOD-loaded thermo-sensitive hydrogel-poly(N-isopropyl-acrylamide)/poly(γ-glutamic acid) | [213] | |
| SOD3-overexpressing MSCs | [214,215] | |
| RM191A | [216] | |
| renal diseases | hEC-SOD tempol | [217,218] [219,220,221], [222] |
| metabolic diseases | SODB | [223,224] |
| nano-SOD | [225,226,227] | |
| MnTE-2-PyP5+ | [228] | |
| ocular diseases | SOD1 | [229,230] |
| rMnSOD | [231] * |
| SOD/SOD Donor | SOD Mimetics | Gene Therapy |
|---|---|---|
| CAR-modified liposomes fasudil plus SOD | [Fe(HPClNOL)Cl2]NO3 | SOD3-overexpressing MSCs |
| gliadin SOD | MnTDE-2-ImP5+ | |
| hEC-SOD | Calmangafodipir * | |
| MS-AOE® | EUK-134 | |
| nano-SOD | EUK-207 | |
| O-HTCC-SOD | GC4419 * | |
| PC-SOD | Nano-MnTnBuOE-2-PyP5+ | |
| rMnSOD * | Mangafodipir * | |
| SOD-loaded thermo-sensitive hydrogel-poly(N-isopropyl-acrylamide)/poly(γ-glutamic acid) | mito-tempo | |
| SOD-loaded porous polymersome | Mn1 | |
| SOD * | MnTE-2-PyP5+ * | |
| SOD1 | MnTM-4-PyP5+ | |
| SOD2 | MnTnBuOE-2-PyP5+ * | |
| SOD2 by Bacillus amyloliquefaciens strain | MnTnHex-2-PyP5+ | |
| SOD3 | RM191A * | |
| SODB * | SOD2m | |
| TAT-SOD * | Tempol * |
| Terrestrial Plants | Microbial | Cyanobacteria | Chromista | Marine Plants |
|---|---|---|---|---|
| Allium cepa L. | Anabaena Geobacillus sp. | Anabaena cylindrica | Lingulodinium polyedrum | Avicennia marina |
| Anacardium occidentale L. | Bacillus amyloliquefaciens | Anabaena variabilis Kutz | Minutocellus polymorphus | Bruguiera gymnorrhiza |
| Camellia sinensis | Bacillus subtilis | Cyanobacterium synechococcus | Nitzschia closterium | Enteromorpha linza |
| Cucumis melo L.C. | Brucella abortus | Microcystis aeruginosa | Thallassiosira weissflogii | Platymonas subcordiformis |
| Cucurbitamoschata L. | Caulobacter crescentus | Nostoc commune | Porphyridium cruentum | |
| Fagopyrum tataricum | Escherichia coli | Nostoc PCC 7120 | Sonneratia alba | |
| Gossypium herbaceum L. | Haemophilus influenzae | Plectonema boryanum | Tetraselmis gracilis | |
| Hordeum vulgare | Haemophilus parainfluenzae | Plectonema boryanum | ||
| Luffa cylindrical | Lactobacillus fermentum | |||
| Momordica charantia | Nodularia Aphanizomenon | |||
| Momordicacharantia L. | Photobacterium leiognathi | |||
| Nicotiana tabacum | Photobacterium phosphoreum | |||
| Olea europaea L. | Photobacterium sepia | |||
| Pisum sativum | Pseudomonas aeruginosa | |||
| Rosmarinus officinalis * | ||||
| Saccharum spp. | ||||
| Salvia officinalis * | ||||
| Syzygium cumini | ||||
| Thymus officinalis * | ||||
| Vitis vinifera L. | ||||
| Zea mays L. |
| Cyclic Polyamines | MnPLED | MnPs | Salen-Mn Complexes | Metal-Based Compounds | Nitroxide |
|---|---|---|---|---|---|
| GC4419 | calmangafodipir | MnTDE-2-ImP5+ | EUK-134 | [Fe(HPClNOL)Cl2]NO3 | mito-tempo |
| Mn1 | mangafodipir | MnTE-2-PyP5+ | EUK-207 | RM191A | tempol |
| MnTM-4-PyP5+ | |||||
| MnTnBuOE-2-PyP5+ | |||||
| MnTnHex-2-PyP5+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosa, A.C.; Corsi, D.; Cavi, N.; Bruni, N.; Dosio, F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules 2021, 26, 1844. https://doi.org/10.3390/molecules26071844
Rosa AC, Corsi D, Cavi N, Bruni N, Dosio F. Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules. 2021; 26(7):1844. https://doi.org/10.3390/molecules26071844
Chicago/Turabian StyleRosa, Arianna Carolina, Daniele Corsi, Niccolò Cavi, Natascia Bruni, and Franco Dosio. 2021. "Superoxide Dismutase Administration: A Review of Proposed Human Uses" Molecules 26, no. 7: 1844. https://doi.org/10.3390/molecules26071844
APA StyleRosa, A. C., Corsi, D., Cavi, N., Bruni, N., & Dosio, F. (2021). Superoxide Dismutase Administration: A Review of Proposed Human Uses. Molecules, 26(7), 1844. https://doi.org/10.3390/molecules26071844