
Synthesis and Biological Screening of New Cyano-Substituted Pyrrole Fused (Iso)Quinoline Derivatives

 , ,
, ,  and
and
Abstract
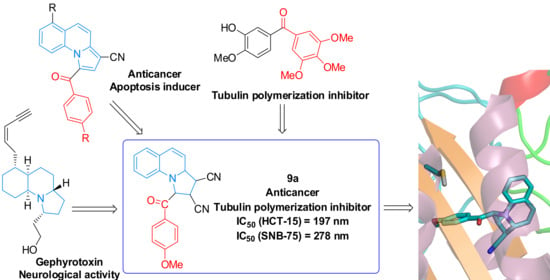
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of Isoquinolinium and Quinolinium Salts 3 and 5
3.1.2. General Procedure for Synthesis of Isoquinoline and Quinoline Derivatives 7, 8, 9 and 10
3.1.3. Spectral Data
2-(2-(4-Cyanophenyl)-2-oxoethyl)isoquinolin-2-ium bromide (3c)
1-(2-(4-cyanophenyl)-2-oxoethyl)quinolin-1-ium bromide 5c
3-(4-Methoxybenzoyl)-1,2,3,10b-tetrahydropyrrolo[2,1-a]isoquinoline-1,2-dicarbonitrile 7a
3-(4-Bromobenzoyl)-1,2,3,10b-tetrahydropyrrolo[2,1-a]isoquinoline-1,2-dicarbonitrile 7b
3-(4-cyanobenzoyl)pyrrolo[2,1-a]isoquinoline-1-carbonitrile 8c
1-(4-methoxybenzoyl)-1,2,3,3a-tetrahydropyrrolo[1,2-a]quinoline-2,3-dicarbonitrile 9a
1-(4-bromobenzoyl)-1,2,3,3a-tetrahydropyrrolo[1,2-a]quinoline-2,3-dicarbonitrile 9b
2-cyano-1-(4-cyanobenzoyl)pyrrolo[1,2-a]quinoline-3-carboxamide 10c
3.2. Anticancer Activity
3.3. Tubulin Polymerization Assay
3.4. Molecular Modelling
3.5. In Silico Prediction of Cytotoxicity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, J.; Gao, P.; Sun, L.; Song, Y.; Kang, D.; Liu, X.; Zhan, P. Efficient drug discovery by rational lead hybridization based on crystallographic overlay. Drug Discov. Today 2019, 24, 805–813. [Google Scholar] [CrossRef]
- Panda, P.; Chakroborty, S. Navigating the Synthesis of Quinoline Hybrid Molecules as Promising Anticancer Agents. ChemistrySelect 2020, 5, 10187–10199. [Google Scholar] [CrossRef]
- Abbot, V.; Sharma, P.; Dhiman, S.; Noolvi, M.N.; Patel, H.M.; Bhardwaj, V. Small hybrid heteroaromatics: Resourceful biological tools in cancer research. RSC Adv. 2017, 7, 28313–28349. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Chandra, V.; Jain, P.K.; Pathak, K.; Pathak, D.; Vaidya, A. Comprehensive review on current developments of quinoline-based anticancer agents. Arab. J. Chem. 2019, 12, 4920–4946. [Google Scholar] [CrossRef] [Green Version]
- Matveeva, M.D.; Purgatorio, R.; Voskressensky, L.G.; Altomare, C.D. Pyrrolo[2,1-a]isoquinoline scaffold in drug discovery: Advances in synthesis and medicinal chemistry. Future Med. Chem. 2019, 11, 2735–2755. [Google Scholar] [CrossRef] [PubMed]
- Pässler, U.; Knölker, H.J. The pyrrolo[2,1-a]isoquinoline alkaloids. Alkaloids Chem. Biol. 2011, 70, 79–151. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.N.; Zeng, K.W.; Zhao, M.B.; Jiang, Y.; Tu, P.F. Pyrrolo[2,1-a]isoquinoline and pyrrole alkaloids from Sinomenium acutum. J. Asian Nat. Prod. Res. 2017, 20, 195–200. [Google Scholar] [CrossRef]
- Bailly, C. Anticancer Properties of Lamellarins. Mar. Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef] [Green Version]
- Marco, E.; Laine, W.; Tardy, C.; Lansiaux, A.; Iwao, M.; Ishibashi, F.; Bailly, C.; Gago, F. Molecular Determinants of Topoisomerase I Poisoning by Lamellarins: Comparison with Camptothecin and Structure-Activity Relationships. J. Med. Chem. 2005, 48, 3796–3807. [Google Scholar] [CrossRef]
- Allin, S.M.; Gaskell, S.N.; Towler, J.M.R.; Bulman Page, P.C.; Saha, B.; McKenzie, M.J.; Martin, W.P. A New Asymmetric Synthesis of the Anti-Tumor Alkaloid (R)-(+)-Crispine A. J. Org. Chem. 2007, 72, 8972–8975. [Google Scholar] [CrossRef]
- Tokuyama, T.; Uenoyama, K.; Brown, G.; Daly, J.W.; Witkop, B. Allenic and Acetylenic Spiropiperidine Alkaloids from the Neotropical Frog, Dendrobates histrionicus. Helv. Chim. Acta 1974, 57, 2597–2604. [Google Scholar] [CrossRef]
- Santarem, M.; Vanucci-Bacqué, C.; Lhommet, G. Formal Total Synthesis of (+)-Gephyrotoxin. J. Org. Chem. 2008, 73, 6466–6469. [Google Scholar] [CrossRef]
- Kemnitzer, W.; Kuemmerle, J.; Jiang, S.; Zhang, H.-Z.; Sirisoma, N.; Kasibhatla, S.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery of 1-benzoyl-3-cyanopyrrolo[1,2-a]quinolines as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. Part 1: Structure-activity relationships of the 1- and 3-positions. Bioorg. Med. Chem. Lett. 2008, 18, 6259–6264. [Google Scholar] [CrossRef]
- Kemnitzer, W.; Kuemmerle, J.; Jiang, S.; Zhang, H.-Z.; Sirisoma, N.; Kasibhatla, S.; Crogan-Grundy, C.; Tseng, B.; Drewe, J.; Cai, S.X. Discovery of 1-benzoyl-3-cyanopyrrolo[1,2-a]quinolines as a new series of apoptosis inducers using a cell- and caspase-based high-throughput screening assay. 2: Structure-activity relationships of the 4-, 5-, 6-, 7- and 8-positions. Bioorg. Med. Chem. Lett. 2009, 19, 3481–3484. [Google Scholar] [CrossRef] [PubMed]
- Brazhko, O.A.; Yevlash, A.S.; Zavgorodnii, M.P.; Kornet, M.M.; Brazhko, O.O.; Lagron, A.V. Basic approaches to the synthesis of pyrrolo[1,2-a]quinolines derivatives: A review. Vopr. Khimii Khimicheskoi Tekhnologii 2019, 6, 6–16. [Google Scholar] [CrossRef]
- Sardaru, M.C.; Crăciun, A.M.; Al Matarneh, C.M.; Sandu, I.A.; Amarandi, R.M.; Popovici, L.; Ciobanu, C.I.; Peptanariu, D.; Pinteala, M.; Mangalagiu, I.I.; et al. Cytotoxic substituted indolizines as new colchicine site tubulin polymerisation inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Popovici, L.; Amarandi, R.M.; Mangalagiu, I.I.; Mangalagiu, V.; Danac, R. Synthesis, molecular modelling and anticancer evaluation of new pyrrolo[1,2-b]pyridazine and pyrrolo[2,1-a]phthalazine derivatives. J. Enzym. Inhib. Med. Chem. 2019, 34, 230–243. [Google Scholar] [CrossRef] [Green Version]
- Al Matarneh, C.M.; Amarandi, R.M.; Craciun, A.M.; Mangalagiu, I.I.; Zbancioc, G.; Danac, R. Design, synthesis, molecular modelling and anticancer activities of new fused phenanthrolines. Molecules 2020, 25, 527–543. [Google Scholar] [CrossRef] [Green Version]
- Danac, R.; Al Matarneh, C.M.; Shova, S.; Daniloaia, T.; Balan, M.; Mangalagiu, I.I. New indolizines with phenanthroline skeleton: Synthesis, structure, antimycobacterial and anticancer evaluation. Bioorg. Med. Chem. 2015, 23, 2318–2327. [Google Scholar] [CrossRef] [PubMed]
- Al Matarneh, C.M.; Shova, S.; Mangalagiu, I.I.; Danac, R. Synthesis, structure and antimycobacterial properties of new pyrrolo-4,7-phenanthroline derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Al Matarneh, C.M.; Ciobanu, C.; Mangalagiu, V.; Zbancioc, G.; Danac, R. Microwave Assisted Synthesis of Six Member Ring Azaheterocycles and Their Antimycobacterial and Anticancer Evaluation. Rev. Chim. 2020, 71, 287–293. [Google Scholar] [CrossRef]
- Fatiadi, A.J. Preparation and Synthetic Applications of Cyano Compounds. In Triple-Bonded Functional Groups: Supplement C: Part 2, Volume 2; Patai, S., Rappaport, Z., Eds.; Wiley: New York, NY, USA, 1983; pp. 1057–1303. [Google Scholar] [CrossRef]
- Al Matarneh, C.M.; Apostu, M.O.; Mangalagiu, I.I.; Danac, R. Reactions of ethyl cyanoformate with cycloimmonium salts: A direct pathway to fused or substituted azaheterocycles. Tetrahedron 2016, 72, 4230–4238. [Google Scholar] [CrossRef]
- Al Matarneh, C.M.; Ciobanu, C.I.; Apostu, M.O.; Mangalagiu, I.I.; Danac, R. Cycloaddition versus amidation in reactions of 2–amino-2-oxoethyl phenanthroliniumylides to activated alkynes and alkenes. C. R. Chem. 2018, 21, 1–8. [Google Scholar] [CrossRef]
- Al Matarneh, C.M.; Isac, D.L.; Tigoianu, R.; Danac, R.; Airinei, A. Steady state and time resolved fluorescence studies of new indolizine with phenanthroline skeleton derivatives. J. Lumin. 2018, 199, 6–12. [Google Scholar] [CrossRef]
- Al Matarneh, C.M.; Danac, R.; Leontie, L.; Tudorache, F.; Petrila, I.; Iacomi, F.; Carlescu, A.; Nedelcu, G.; Mangalagiu, I. Synthesis and electron transport properties of some new 4,7-phenanthroline derivatives in thin films. Environ. Eng. Manag. J. 2015, 14, 420–431. [Google Scholar]
- Al Matarneh, C.M.; Ciobanu, C.I.; Mangalagiu, I.I.; Danac, R. Design, synthesis and antimycobacterial activity of some new azaheterocycles: 4,7-phenanthroline with p-halogeno-benzoyl skeleton. Part VI. J. Serb. Chem. Soc. 2016, 81, 133–140. [Google Scholar] [CrossRef]
- Al Matarneh, C.M.; Sardaru, M.C.; Apostu, M.O.; Rosca, I.; Ciobanu, C.I.; Mangalagiu, I.I.; Danac, R. Synthesis and antibacterial evaluation of new pyrrolo [3′,4′:3,4]pyrrolo[1,2-a]Quinoline and pyrrolo[3′,4′:3,4]pyrrolo[2,1-a]Isoquinoline derivatives. Studia UBB Chem. 2019, 64, 67–80. [Google Scholar] [CrossRef]
- Hopkin, M.D.; Baxendale, I.R.; Ley, S.V. A New Focused Microwave Approach to the Synthesis of Amino-Substituted Pyrroloisoquinolines and Pyrroloquinolines via a Sequential Multi-Component Coupling Process. Synthesis 2008, 11, 1688–1702. [Google Scholar] [CrossRef] [Green Version]
- Jasinski, R.; Dresler, E. On the Question of Zwitterionic Intermediates in the [3+2] Cycloaddition Reactions: A Critical Review. Organics 2020, 1, 49–69. [Google Scholar] [CrossRef]
- Mloston, G.; Urbaniak, K.; Domagała, M. Thermal [2+3]-Cycloadditions of trans-1-Methyl-2,3-diphenylaziridine with C=S and C=C Dipolarophiles: An Unexpected Course with Dimethyl Dicyanofumarate. Helv. Chim. Acta 2009, 92, 2631–2642. [Google Scholar] [CrossRef]
- Huisgen, R.; Mloston, G.; Langhals, E. The first two-step 1,3-dipolar cycloadditions: Interception of intermediate. J. Org. Chem. 1986, 51, 4085–4087. [Google Scholar] [CrossRef]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesh, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Boyd, R.B. The NCI in vitro anticancer drug discovery screen: Concept, implementation, and operation. In Anticancer Drug Development Guide: Preclinical Screening, Clinical Trials, and Aproval; Teicher, B., Ed.; Humana Press Inc.: Totowa, NJ, USA, 1997; pp. 23–42. [Google Scholar]
- Lagunin, A.A.; Dubovskaja, V.I.; Rudik, A.V.; Pogodin, P.V.; Druzhilovskiy, D.S.; Gloriozova, T.A.; Filimonov, D.A.; Sastry, N.G.; Poroikov, V.V. CLC-Pred: A freely available web-service for in silico prediction of human cell line cytotoxicity for drug-like compounds. PLoS ONE 2018, 12, e0191838. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, J.; Wang, J.; Li, C.M.; Ahn, S.; Barrett, C.M.; Dalton, J.T.; Li, W.; Miller, D.D. Design, synthesis, and biological evaluation of stable colchicine binding site tubulin inhibitors as potential anticancer agents. J. Med. Chem. 2014, 57, 7355–7366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klejborowska, G.; Urbaniak, A.; Maj, E.; Pretod, J.; Mosharie, M.; Wietrzykc, J.; Tuszynskie, J.A.; Chambers, T.C.; Huczyński, A. Synthesis, biological evaluation and molecular docking studies of new amides of 4-chlorothiocolchicine as anticancer agents. Bioorg. Chem. 2020, 97, 103664–103675. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.; Shokrzadeh, M.; Karima, S.; Rajaei, S.; Hashemi, S.M.; Mirzaei, H.; Fallah, M.; Emami, S. Design, synthesis and biological evaluation of flexible and rigid analogs of 4H-1,2,4-triazoles bearing 3,4,5-trimethoxyphenyl moiety as new antiproliferative agents. Bioorg. Chem. 2019, 93, 103300–103312. [Google Scholar] [CrossRef] [PubMed]
- Onodera, K.; Satou, K.; Hirota, H. Evaluations of Molecular Docking Programs for Virtual Screening. J. Chem. Inf. Model. 2007, 47, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Schofield, A.V.; Gamell, C.; Suryadinata, R.; Sarcevic, B.; Bernard, O. Tubulin polymerization promoting protein 1 (Tppp1) phosphorylation by Rho-associated coiled-coil kinase (Rock) and cyclin-dependent kinase 1 (Cdk1) inhibits microtubule dynamics to increase cell proliferation. J. Biol. Chem. 2013, 288, 7907–7917. [Google Scholar] [CrossRef] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDockVina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, A.E.; Danel, F.; Bachmann, F.; Bargsten, K.; Buey, R.M.; Pohlmann, J.; Reinelt, S.; Lane, H.; Steinmetz, M.O. The novel microtubule-destabilizing drug BAL27862 binds to the colchicine site of tubulin with distinct effects on microtubule organization. J. Mol. Biol. 2014, 426, 1848–1860. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | H1 δ(ppm) | J1,2 (Hz) | H2 δ(ppm) | J2,3 (Hz) | H3 δ(ppm) | J3,10 (Hz) | H10 δ(ppm) |
|---|---|---|---|---|---|---|---|
| 7a | 6.04 | 2.50 | 4.26 | 8.00 | 3.79 | 9.50 | 4.32 |
| 7b | 6.00 | 8.00 | 4.33 | 4.50 | 4.40 | 6.50 | 5.04 |
| Compound | H1 δ(ppm) | J1,2 (Hz) | H2 δ(ppm) | J2,3 (Hz) | H3 δ(ppm) | J3,10 (Hz) | H10 δ(ppm) |
|---|---|---|---|---|---|---|---|
| 9a | 5.89 | 9.00 | 4.72 | 4.00 | 4.21 | - | 5.12 |
| 9b | 5.96 | 9.00 | 4.72 | 4.00 | 4.22 | 2.00 | 5.10 |
| Cell Type | Cell Line | Compound | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | 3b | 3c | 5a | 5b | 5c | 7a | 7b | 8c | 9a | 9b | Phenstatin | ||
| Leukemia | CCRF-CEM | 8 | 6 | 2 | 1 | 0 | 0 | 18 | 0 | 0 | 84 | 15 | 94 |
| K-562 | 4 | 20 | 0 | 11 | 17 | 7 | 27 | 0 | 12 | 88 | 76 | 91 | |
| SR | 0 | 6 | 0 | 0 | 0 | 3 | 0 | 0 | 25 | 79 | 75 | 93 | |
| HL-60(TB) | 0 | 14 | 0 | 2 | 0 | 1 | 6 | 0 | 8 | 100 b,d | 6 | 100 b,l | |
| MOLT-4 | 3 | 7 | 2 | 0 | 0 | 0 | 5 | 0 | 7 | 63 | 8 | 85 | |
| RPMI-8226 | 0 | 6 | 3 | 0 | 1 | 0 | 27 | 0 | 0 | 70 | 6 | 87 | |
| Non-small Cell Lung Cancer | A549/ATCC | 0 | 3 | 1 | 2 | 2 | 0 | 13 | 17 | 10 | 70 | 35 | 82 |
| HOP-92 | 18 | 31 | 24 | 15 | 24 | 14 | 15 | 5 | 20 | 16 | 45 | 48 | |
| HOP-62 | 10 | 18 | 9 | 3 | 13 | 0 | 7 | 3 | 66 | 70 | 25 | 77 | |
| NCI-H460 | 0 | 3 | 0 | 0 | 0 | 0 | 3 | 13 | 28 | 81 | 4 | 93 | |
| NCI-H522 | 8 | 11 | 2 | 8 | 6 | 4 | 29 | 12 | 36 | 100 | 22 | 88 | |
| Colon Cancer | COLO205 | 1 | 18 | 8 | 0 | 0 | 0 | 0 | 0 | 0 | 100 b,e | 97 | 58 |
| HCT-116 | 3 | 27 | 11 | 4 | 5 | 2 | 14 | 13 | 15 | 85 | 32 | 96 | |
| HCT-15 | 2 | 5 | 2 | 8 | 1 | 0 | 5 | 4 | 5 | 83 | 77 | 96 | |
| HT-29 | 5 | 33 | 0 | 3 | 2 | 5 | 16 | 14 | 20 | 93 | 63 | 85 | |
| SW-620 | 7 | 13 | 9 | 5 | 8 | 4 | 82 | 0 | 8 | 74 | 34 | 78 | |
| KM12 | 6 | 10 | 1 | 1 | 2 | 4 | 2 | 0 | 10 | 94 | 78 | 91 | |
| CNS Cancer | SF-295 | 3 | 17 | 2 | 7 | 5 | 5 | 5 | 4 | 36 | 78 | 11 | 100 b,m |
| SF-539 | 1 | 8 | 5 | 0 | 0 | 2 | 5 | 11 | 52 | 100 b,f | 11 | 100 b,n | |
| SNB-75 | 20 | 24 | 30 | 29 | 29 | 17 | 26 | 10 | 67 | 100 b,f | 88 | 100 b,o | |
| U251 | 0 | 1 | 2 | 3 | 0 | 0 | 7 | 0 | 15 | 66 | 16 | 79 | |
| Melanoma | LOX IMVI | 5 | 14 | 2 | 6 | 7 | 8 | 13 | 8 | 20 | 57 | 22 | 85 |
| M14 | 5 | 13 | 8 | 7 | 6 | 5 | 6 | 0 | 3 | 100 b,g | 25 | 100 b,p | |
| MDA-MB-435 | 0 | 8 | 2 | 0 | 2 | 0 | 4 | 0 | 0 | 100 b,h | 84 | 100 b,r | |
| UACC-62 | 14 | 18 | 0 | 11 | 11 | 13 | 37 | 2 | 35 | 53 | 37 | 55 | |
| SK-MEL-2 | 0 | 3 | 0 | 0 | 0 | 0 | 12 | 2 | 7 | 66 | 0 | 40 | |
| SK-MEL-5 | 0 | 2 | 0 | 0 | 1 | 0 | 14 | 10 | 11 | 85 | 39 | 100 b,s | |
| Ovarian Cancer | OVCAR-3 | 3 | 21 | 0 | 0 | 3 | 0 | 8 | 0 | 8 | 100 b,i | 25 | 100 b,g |
| NCI/ADR-RES | 3 | 6 | 2 | 2 | 0 | 0 | 13 | 6 | 45 | 100 b,d | 36 | 100 b,t | |
| SK-OV-3 | 3 | 19 | 4 | 0 | 18 | 1 | 7 | 0 | 0 | 45 | 21 | 53 | |
| OVCAR-4 | 3 | 17 | 4 | 0 | 9 | 0 | 38 | 46 | 57 | 54 | 57 | 69 | |
| OVCAR-8 | 3 | 2 | 5 | 2 | 3 | 0 | 100 b,c | 6 | 58 | 73 | 10 | 86 | |
| Renal cancer | A498 | 7 | 11 | 56 | 0 | 5 | 3 | 20 | 0 | 5 | 100 b,j | 33 | 25 |
| RXF393 | 1 | 7 | 0 | 0 | 1 | 0 | 23 | 0 | 25 | 100 b,k | 25 | 99 | |
| UO-31 | 19 | 29 | 17 | 14 | 20 | 18 | 40 | 1 | 16 | 68 | 52 | 67 | |
| Breast cancer | MCF7 | 8 | 14 | 11 | 11 | 17 | 10 | 9 | 8 | 17 | 92 | 87 | 94 |
| MDA-MB-468 | 10 | 16 | 10 | 4 | 9 | 15 | 36 | 0 | 23 | 84 | 100 b,d | 100 b,u | |
| HS 578T | 5 | 9 | 6 | 10 | 8 | 5 | 5 | 0 | 50 | 86 | 23 | 71 | |
| BT-549 | 0 | 0 | 5 | 0 | 0 | 0 | 5 | 0 | 47 | 47 | 7 | 88 | |
| Prostate cancer | PC-3 | 11 | 18 | 8 | 10 | 13 | 9 | 16 | 6 | 39 | 71 | 24 | 80 |
| DU-145 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 | 18 | 76 | 9 | 90 | |
| Cell Type | Compound | 9a |
|---|---|---|
| Cell Line | GI50 (μM) b | |
| Leukemia | HL-60(TB) | 1.23 |
| SR | 0.544 | |
| K-562 | 0.405 | |
| Non-small Cell Lung Cancer | NCI-H460 | 0.749 |
| NCI-H522 | 2.45 | |
| A549/ATCC | 2.40 | |
| Colon Cancer | HCT-116 | 0.653 |
| HCT-15 | 0.197 | |
| HT29 | 0.844 | |
| SW-620 | 0.715 | |
| KM12 | 1.25 | |
| Prostate cancer | PC-3 | 1.45 |
| DU-145 | 2.93 | |
| Melanoma | SK-MEL-5 | 3.49 |
| M14 | 0.50 | |
| MDA-MB-435 | 0.335 | |
| UACC-62 | 0.781 | |
| LOXIMVI | 2.10 | |
| MALME-3M | 2.67 | |
| Cell line | GI50 (μM) b | |
| Ovarian Cancer | OVCAR-3 | 0.642 |
| NCI/ADR-RES | 0.572 | |
| IGROV1 | 2.74 | |
| OVCAR-8 | 3.29 | |
| Renal Cancer | CAKI-1 | 0.460 |
| A498 | 0.244 | |
| UO-31 | 1.95 | |
| ACHN | 1.43 | |
| RXF 393 | 1.52 | |
| Breast cancer | MCF7 | 1.09 |
| HS 578T | 2.10 | |
| MDA-MB-231/ATCC | 1.46 | |
| BT-549 | 1.98 | |
| T-47D | 2.39 | |
| MDA-MB-468 | 2.31 | |
| CNS Cancer | SF-295 | 2.83 |
| SF-539 | 1.32 | |
| SNB-75 | 0.278 | |
| U251 | 2.81 |
| Pa | Pi | Cell Line | Cell Type |
|---|---|---|---|
| 0.698 | 0.009 | DU-145 | Prostate carcinoma |
| 0.521 | 0.004 | A-375 | Malignant melanoma |
| 0.500 | 0.029 | PC-3 | Prostate carcinoma |
| 0.438 | 0.035 | A2058 | Melanoma |
| 0.308 | 0.007 | LNCaP | Prostate carcinoma |
| 0.399 | 0.100 | YAPC | Pancreatic carcinoma |
| 0.391 | 0.125 | Hs 683 | Oligodendroglioma |
| 0.340 | 0.080 | U-266 | Plasma cell myeloma |
| 0.323 | 0.214 | NALM-6 | Adult B acute lymphoblastic leukemia |
| 0.329 | 0.249 | MDA-MB-453 | Breast adenocarcinoma |
| Compound | Binding Energy (kcal/mol) | Binding Orientation | 2D Interaction Diagram |
|---|---|---|---|
| Phen | −7.7 |  |  |
| 3a | −8.7 |  |  |
| 3b | −8.6 |  |  |
| 3c | −9.2 |  |  |
| 5a | −8.6 |  |  |
| 5b | −8.6 |  |  |
| 5c | −9.3 |  |  |
| 7a | −9.7 |  |  |
| 7b | −9.4 |  |  |
| 8c | −10.8 |  |  |
| 9a | −9.8 |  |  |
| 9b | −10.1 |  |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Matarneh, M.C.; Amărandi, R.-M.; Mangalagiu, I.I.; Danac, R. Synthesis and Biological Screening of New Cyano-Substituted Pyrrole Fused (Iso)Quinoline Derivatives. Molecules 2021, 26, 2066. https://doi.org/10.3390/molecules26072066
Al-Matarneh MC, Amărandi R-M, Mangalagiu II, Danac R. Synthesis and Biological Screening of New Cyano-Substituted Pyrrole Fused (Iso)Quinoline Derivatives. Molecules. 2021; 26(7):2066. https://doi.org/10.3390/molecules26072066
Chicago/Turabian StyleAl-Matarneh, Maria Cristina, Roxana-Maria Amărandi, Ionel I. Mangalagiu, and Ramona Danac. 2021. "Synthesis and Biological Screening of New Cyano-Substituted Pyrrole Fused (Iso)Quinoline Derivatives" Molecules 26, no. 7: 2066. https://doi.org/10.3390/molecules26072066
APA StyleAl-Matarneh, M. C., Amărandi, R. -M., Mangalagiu, I. I., & Danac, R. (2021). Synthesis and Biological Screening of New Cyano-Substituted Pyrrole Fused (Iso)Quinoline Derivatives. Molecules, 26(7), 2066. https://doi.org/10.3390/molecules26072066