
Development and Validation of a Chiral Liquid Chromatographic Assay for Enantiomeric Separation and Quantification of Verapamil in Rat Plasma: Stereoselective Pharmacokinetic Application

 , , ,
, , , 
Abstract
:1. Introduction
2. Results and Discussion
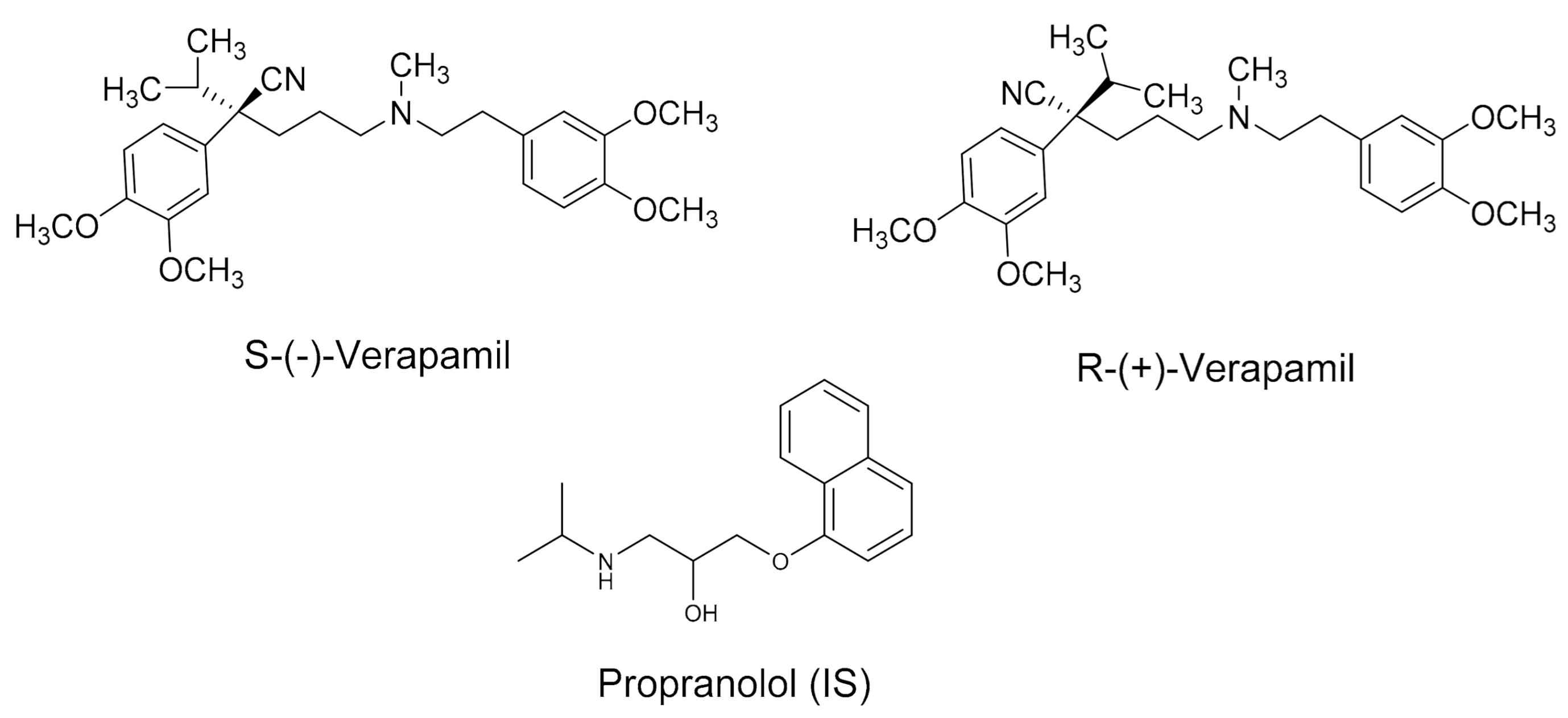
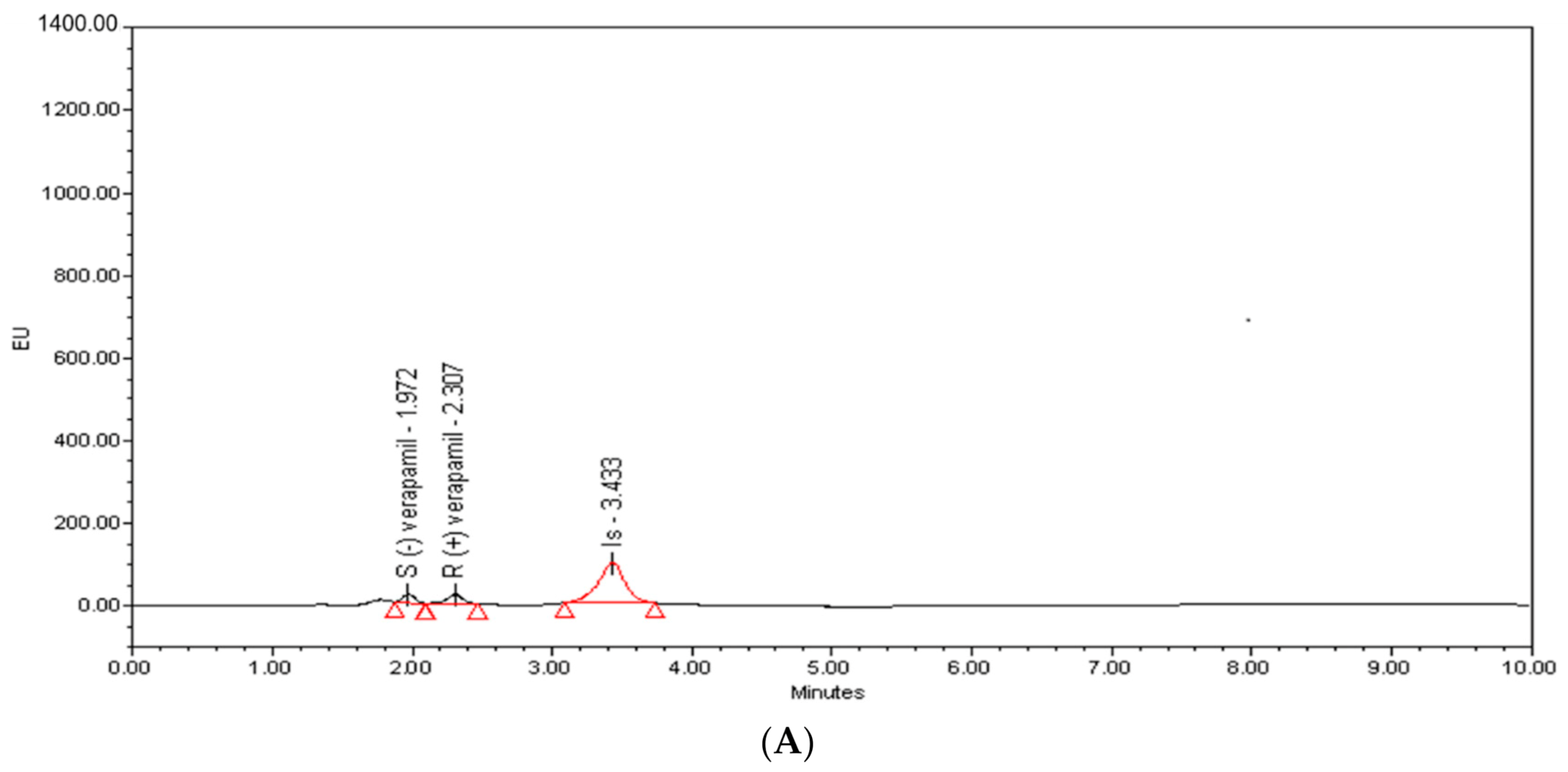
2.1. Method Development and Optimization
2.2. Sample Pretreatment
2.3. In-Study Validation
2.4. Application to an Enantioselective Pharmacokinetic Study
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Chromatographic Conditions
3.3. Animals
3.4. Sample Preparation
3.4.1. Preparation of Stock and Standard Solutions
3.4.2. Preparation of Calibrators and Quality Control Samples
3.5. Sample Pretreatment
3.6. Pre-Study Validation
3.7. Stereoselective Pharmacokinetic Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ibrahim, A.; Hashem, H.; Elhenawee, M.; Saleh, H. Comparison between core-shell and totally porous particle stationary phases for fast and green LC determination of five hepatitis-C antiviral drugs. J. Sep. Sci. 2018, 41, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Avela, H.; Siren, H. Advances in analytical tools and current statistical methods used in ultra-high-performance liquid chromatography-mass spectrometry of glycero-, glycerophospho- and sphingolipids. Int. J. Mass Spectrom. 2020, 457, 116408. [Google Scholar] [CrossRef]
- Lesko, M.; Samuelsson, J.; Asberg, D.; Kaczmarskib, K.; Fornstedt, T. Evaluating the advantages of higher heat conductivity in a recently developed type of core-shell diamond stationary phase particle in UHPLC. J. Chromatogr. A 2020, 1625, 461076. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Armstrong, D.W. Fast super/subcritical fluid chromatographic enantioseparations on superficially porous particles bonded with broad selectivity chiral selectors relative to fully porous particles. J. Chromatogr. A 2019, 1605, 360339. [Google Scholar] [CrossRef]
- Gumustas, M.; Zalewski, P.; Ozkan, S.; Uslu, B. The history of the core–shell particles and applications in active pharmaceutical ingredients via liquid chromatography. Chromatographia 2019, 82, 17–48. [Google Scholar] [CrossRef]
- Broeckhoven, K.; Cabooter, D.; Desmet, G. Kinetic performance comparison of fully and superficially porous particles with sizes ranging between 2.7 µm and 5 μm: Intrinsic evaluation and application to a pharmaceutical test compound. J. Pharm. Anal. 2013, 3, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, N.; McCalley, D.V. Core-shell, ultra small particles, monoliths and other support materials in HPLC. Anal. Chem. 2015, 88, 279–298. [Google Scholar] [CrossRef]
- Hetzel, T.; Loeker, D.; Teutenberg, T.; Schmidt, T.C. Characterization of the efficiency of microbore LC columns by Van Deemter and kinetic plot analysis. J. Sep. Sci. 2016, 39, 3889–3897. [Google Scholar] [CrossRef]
- Urio, R.P.; Masini, J.C. Evaluation of monolithic and core-shell columns for separation of triazine herbicides by reversed phase HPLC. J. Braz. Chem. Soc. 2015, 26, 2331–2338. [Google Scholar]
- Elkurdi, S.; Abumuaileq, D.; Alhazmi, H.; Albratty, M.; Eldeeb, A. Comparing monolithic and fused core HPLC columns for fast chromatographic analysis of fat-soluble vitamins. Acta Pharm. 2017, 67, 203–213. [Google Scholar]
- Barhate, C.L.; Lopez, D.A.; Makarov, A.A.; Bu, X.; Morris, W.J.; Lekhal, A.; Hartman, R.; Armstrong, D.W.; Regalado, E.L. Macrocyclic glycopeptide chiral selectors bonded to core-shell particles enables enantiopurity analysis of the entire verubecestat synthetic route. J. Chromatogr. A 2018, 1539, 87–92. [Google Scholar] [CrossRef]
- Hellinghausen, G.; Roy, D.; Lee, J.T.; Wang, Y.; Weatherly, C.A.; Lopez, D.A.; Nguyen, K.A.; Armstrong, J.D.; Armstrong, D.W. Effective methodologies for enantiomeric separations of 150 pharmacology and toxicology related 1°, 2°, and 3° amines with core-shell chiral stationary phases. J. Pharm. Biomed. Anal. 2018, 155, 70–81. [Google Scholar] [CrossRef]
- Patel, D.C.; Breitbach, Z.S.; Yu, J.; Nguyen, K.A.; Armstrong, D.W. Quinine bonded to superficially porous particles for high-efficiency and ultrafast liquid and supercritical fluid chromatography. Anal. Chim. Acta 2017, 963, 164–174. [Google Scholar] [CrossRef]
- Patel, D.C.; Wahab, M.F.; Armstrong, D.W.; Breitbach, Z.S. Advances in high-throughput and high-efficiency chiral liquid chromatographic separations. J. Chromatogr. A 2016, 1467, 2–18. [Google Scholar] [CrossRef]
- Spudeit, D.A.; Dolzan, M.D.; Breitbach, Z.S.; Barber, W.E.; Micke, G.A.; Armstrong, D.W. Superficially porous particles vs. fully porous particles for bonding high performance liquid chromatographic chiral stationary phases: Isopropyl cyclofructan 6. J. Chromatogr. A 2014, 1363, 89–95. [Google Scholar] [CrossRef]
- Busse, D.; Templin, S.; Mikus, G.; Schwab, M.; Hofmann, U.; Eichelbaum, M.; Kivistö, K.T. Cardiovascular effects of (R)—and (S)-verapamil and racemic verapamil in humans: A placebo-controlled study. Eur. J. Clin. Pharmcol. 2006, 62, 613–619. [Google Scholar] [CrossRef]
- Pagel, P.S.; Hettrick, D.A.; Lowe, D.; Gowrie, P.W.; Kersten, J.R.; Bosnjak, Z.J.; Warltier, D.C. Cardiovascular effects of verapamil enantiomer combinations in conscious dogs. Eur. J. Pharmacol. 1998, 348, 213–221. [Google Scholar] [CrossRef]
- Echizen, H.; Manz, M.; Eichelbaum, M. Electrophysiologic effects of dextro—and levo-verapamil on sinus node and AV node function in humans. J. Cardiovasc. Pharmacol. 1988, 12, 543–546. [Google Scholar] [CrossRef]
- Bhatti, M.M.; Foste, R.T. Pharmacokinetics of the enantiomers of verapamil after intravenous and oral administration of racemic verapamil in a rat model. Biopharm. Drug Dispos. 1997, 18, 387–396. [Google Scholar] [CrossRef]
- Regenthal, R.; Krueger, M.; Koeppel, C.; Preiss, R. Drug levels: Therapeutic and toxic serum/plasma concentrations of common drugs. J. Clin. Monit. Comput. 1999, 15, 529–544. [Google Scholar] [CrossRef]
- Eisenberg, J.N.; Oakley, G.D. Probable adverse interaction between oral metoprolol and verapamil. Postgrad. Med. J. 1984, 60, 705–796. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Lv, C.; Zeng, S. Significance and challenges of stereoselectivity assessing methods in drug metabolism. J. Pharm. Anal. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.C.; Saville, D.J.; Wanwimolruk, S. Simultaneous HPLC analysis of S—and R- verapamil and metabolites, S—and R- norverapamil in human plasma. J. Liq. Chrom. Rel. Technol. 2000, 23, 1711–1723. [Google Scholar] [CrossRef]
- Asafu-Adjaye, E.B.; Shiu, G.K. Solid-phase extraction–high-perfomance liquid chromatography determination of verapamil and norverapamil enantiomers in urine. J. Chromatogr. B 1998, 707, 161–167. [Google Scholar] [CrossRef]
- Hanada, K.; Akimoto, S.; Mitsui, K.; Hashiguchi, M.; Ogata, H. Quantitative determination of disopyramide, verapamil and flecainide enantiomers in rat plasma and tissues by high performance liquid chromatography. J. Chromatogr. B 1998, 710, 129–135. [Google Scholar] [CrossRef]
- Fieger, H.; Blaschke, G. Direct determination of the enantiomeric ratio of verapamil, its major metabolite norverapamil and gallopamil in plasma by chiral high-performance liquid chromatography. J. Chromatogr. Biomed. Appl. 1992, 572, 255–260. [Google Scholar] [CrossRef]
- Shibukawa, A.; Wainer, I.W. Simultaneous direct determination of the enantiomers of verapamil and norverapamil in plasma using a derivatized amylose high-performance liquid chromatographic chiral stationary phase. J. Chromatogr. Biomed. Appl. 1992, 574, 85–92. [Google Scholar] [CrossRef]
- Miller, L.; Bergeron, R. Analytical and preparative resolution of enantiomers of verapamil and norverapamil using a cellulose-based chiral stationary phase in the reversed-phase mode. J. Chromatogr. 1993, 648, 381–388. [Google Scholar] [CrossRef]
- Berthod, A.; Jin, H.L.; Beesley, T.E.; Duncan, J.; Armstrongs, D.W. Cyclodextrin chiral stationary phases for liquid chromatographic separations of drug stereoisomers. J. Pharm. Biomed. Anal. 1990, 8, 123–130. [Google Scholar] [CrossRef]
- Brandsteterova, E.; Wainer, I.W. A chiral and chiral high-performance liquid chromatography of verapamil and its metabolites in serum samples. J. Chromatogr. 1999, 732, 395–404. [Google Scholar] [CrossRef]
- Stagni, G.; Gillespie, W.R. Simultaneous analysis of verapamil and norverapamil enantiomers in human plasma by high performance liquid chromatography. J. Chromatogr. B 1995, 667, 349–354. [Google Scholar] [CrossRef]
- Rasymasi, A.K.; Botjdoulas, H.; Kichan, J.M. Determination of verapamil enantiomers in serum following racemate administration using HPLC. J. Liq. Chromatogr. Rel.Tech. 1992, 15, 3013–3029. [Google Scholar] [CrossRef]
- Chu, Y.; Wainer, I.W. Determination of the enantiomers of verapamil and norverapamil in serum using coupled achiral-chiral high-performance liquid chromatography. J. Chromatogr. Biomed. Appl. 1989, 497, 191–200. [Google Scholar] [CrossRef]
- Sandstrom, R.; Lennernas, H.; Ohlen, K.; Karlsson, A. Enantiomeric separation of verapamil and norverapamil using Chiral-AGP as the stationary phase. J. Pharm. Biomed. Anal. 1999, 2, 43–49. [Google Scholar] [CrossRef]
- Lankford, S.M.; Bai, S.A. Determination of the stereochemical composition of the major metabolites of verapamil in doge urine with enantioselective liquid chromatographic techniques. J. Chromatogr. B 1995, 663, 91–101. [Google Scholar] [CrossRef]
- Oda, Y.; Asakawa, N.; Kajima, T.; Yoshida, Y.; Sato, T. On-line determination and resolution of verapamil enantiomers by high-performance liquid chromatography with column switching. J. Chromatogr. 1991, 541, 411–418. [Google Scholar] [CrossRef]
- Hamidia, S.; Jouyban, A. Capillary electrophoresis with UV detection, on-line stacking and off-line dispersive liquid-liquid microextraction for determination of verapamil enantiomers in plasma. Anal. Methods 2015, 7, 5820–5829. [Google Scholar] [CrossRef]
- Resztak, M.; Glowka, F.K. Stereoselective CZE method for analysis of verapamil and norverapamil in human plasma. Acta Pol. Pharm. 2013, 70, 395–401. [Google Scholar] [PubMed]
- Dethy, J.M.; Broux, S.D.; Lesne, M.; Longstreth, J.; Gilbert, P. Stereoselective determination of verapamil and norverapamil by capillary electrophoresis. J. Chromatogr. B 1994, 654, 121–127. [Google Scholar] [CrossRef]
- Ohara, T.; Shibukawa, A.; Nakagawa, T. Capillary electrophoresis/frontal analysis for microanalysis of enantioseiective protein binding of a basic drug. Anal. Chem. 1995, 67, 3520–3525. [Google Scholar] [CrossRef]
- Chankvetadze, B.; Burjanadze, N.; Pintore, G.; Strickmann, D.; Bergenthal, D.; Blaschke, G. Chiral recognition of verapamil by cyclodextrins studied with capillary electrophoresis, NMR spectroscopy, and electrospray ionization mass spectrometry. Chirality 1999, 11, 635–644. [Google Scholar] [CrossRef]
- Stalcup, A.M.; Gahm, K.H. Application of sulfated cyclodextrins to chiral separations by capillary zone electrophoresis. Anal. Chem. 1996, 68, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Clothier, J.G.; Sterling, J.; Tomellini, A. Chiral separation of verapamil and related compounds using micellar electrokinetic capillary chromatography with mixed micelles of bile salt and polyoxyethylene ethers. J. Chromatogr. A 1996, 723, 179–187. [Google Scholar] [CrossRef]
- Todoroki, K.; Kudoh, Y.; Nakamura, M.; Shimizu, Y.; Sasaki, T.; Otsuki, H.; Wada, K.; Min, J.Z.; Mizuno, H.; Yoshinari, K.; et al. Sensitive and comprehensive LC-MS/MS analyses of chiral pharmaceuticals and their hepatic metabolites using ovomucoid column. Anal. Sci. 2018, 34, 1011–1015. [Google Scholar] [CrossRef] [Green Version]
- Singhal, P.; Yadav, M.; Winter, S.; Guttikar, S.; Patel, D.; Mills, M.; Shrivastav, P.S. Enantiomeric separation of verapamil and its active metabolite, norverapamil, and simultaneous quantification in human plasma by LC–ESI-MS-MS. J. Chromatogr. Sci. 2012, 50, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Mateus, F.H.; Lepera, J.S.; Marques, M.P.; Boralli, V.B.; Lanchote, V.L. Simultaneous analysis of the enantiomers of verapamil and norverapamil in rat plasma by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2007, 45, 762–768. [Google Scholar] [CrossRef]
- Alebic-Kolbah, T.; Zavitsanos, A.P. Chiral bioanalysis by normal phase high-performance liquid chromatography-atmospheric pressure ionization tandem mass spectrometry. J. Chromatogr. A 1997, 759, 65–77. [Google Scholar] [CrossRef]
- Hedeland, M.; Fredriksson, E.; Lennernäs, H.; Bondesson, U. Simultaneous quantification of the enantiomers of verapamil and its N-demethylated metabolite in human plasma using liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2004, 804, 303–311. [Google Scholar] [CrossRef]
- Rustichelli, C.; Ferioli, V.; Gamberini, G. Resolution of the enantiomers of verapamil and gallopamil by chiral liquid chromatography-mass spectrometry. Chromatographia 1997, 44, 477–483. [Google Scholar] [CrossRef]
- Declerck, S.; Vander Heyden, Y.; Mangelings, D. Enantioseparations of pharmaceuticals with capillary electrochromatography. A review. J. Pharm. Biomed. Anal. 2016, 130, 81–99. [Google Scholar] [CrossRef]
- Sandra, P. Resolution–definition and nomenclature. J. High. Resolut. Chromatogr. 1989, 12, 82–86. [Google Scholar] [CrossRef]
- Nováková, L.; Vlcková, H. A review of current trends and advances in modern bio-analytical methods: Chromatography and sample preparation. Anal. Chim. Acta 2009, 656, 8–35. [Google Scholar] [CrossRef]
- U. S. Food and Drug Administration. Bioanalytical Method Validation. Guidance for Industry; FDA: Silver Spring, MD, USA, 2018.
- Hu, N.; Xie, S.; Liu, L.; Wang, X.; Pan, X.; Chen, G.; Zhang, L.; Liu, H.; Liu, X.; Liu, X.; et al. Opposite effect of diabetes mellitus induced by streptozotocin on oral and intravenous pharmacokinetics of verapamil in rats. Drug Metab. Dispos. 2011, 39, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.A.; Mehvar, R. Enantioselective distribution of verapamil and norverapamil into human and rat erythrocytes: The role of plasma protein binding. Biopharm. Drug. Dispos. 1996, 17, 577–587. [Google Scholar] [CrossRef]
- Ramaswamy, M.; Wallace, T.L.; Cossum, P.A.; Wasan, K.M. Species differences in the proportion of plasma lipoprotein lipid carried by high-density lipoproteins influence the distribution of free and liposomal nystatin in human, dog, and rat plasma. Antimicrob. Agents Chemother. 1999, 43, 424–1428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Determinate Method | Chiral Selector | Composition of the Mobile Phase | Matrix | Ext. Method | Run Times (min) | LR (ng/mL) | Ref. |
|---|---|---|---|---|---|---|---|
| HPLC-Fluorescence | Chiral-AGP column (α1-acid glycoprotein) (100 mm × 4.0 mm, 5 µm). | Acetonitrile: 10 mM sodium perchlorate, pH 7.0 (10:90, v/v) | Human serum | SPE | 40 | 20–400 | [30] |
| HPLC-Fluorescence | Chiral-AGP column (α1-acid glycoprotein) (100 mm × 4.0 mm, 5 µm). | Acetonitrile: 10 mM sodium perchlorate, pH 7.0 (10:90, v/v) | Human serum | LLE | 40 | 20–400 | [31] |
| HPLC-Fluorescence | Chiralcel OD-RH column (cellulose tris(3,5-di- methylphenylcarbamate) (150 mm × 4.6 mm, 5 µm) | Acetonitrile: 30 mM hexafluorophosphate, pH 4.6 (34:66, v/v) | Human plasma | LLE | 34 | 10–250 | [23] |
| HPLC-Fluorescence | Chiralcel OD-RH column (cellulose tris(3,5-di- methylphenylcarbamate) (250 mm × 4.6 mm, 10 µm) | Acetonitrile: 0.2 M sodium perchlorate (40:60, v/v) | Urine | SPE | 25 | 2.5–300 | [24] |
| HPLC-Fluorescence | Chiralpak AD column (amylose tris(3,5-di- methylphenylcarbamate) (250 mm × 4.6 mm, 5 µm) | n-hexane: isopropanol: diethylamine (94:6:0.1) (v/v/v) | Rat plasma and tissues | LLE | 20 | 100–500 | [25] |
| HPLC-Fluorescence | Chiralpak AD column (amylose tris(3,5-di- methylphenylcarbamate) (250 mm × 4.6 mm, 5 µm | n-hexane: isopropanol: diethylamine (90:10:0.1) (v/v/v) | Human plasma | LLE | 20 | 50–500 | [26] |
| HPLC-Fluorescence | Chiralpak AD column (amylose tris(3,5-di- methylphenylcarbamate) (250 mm × 4.6 mm, 5 µm | n-hexane: isopropanol: triethylamine (85:15:0.4) (v/v/v) | Human plasma | LLE | 20 | 2.5–100 | [27] |
| HPLC-Fluorescence | Chiral-AGP column (α1-acid glycoprotein) (100 mm × 4.0 mm, 5 µm). | Acetonitrile: 30 mM phosphate buffer, pH 5.3 (4:96, v/v) | Human serum | LLE | 20 | 10–120 | [33] |
| Analyte | Rs a | α b | k c | TR (min) d,e |
|---|---|---|---|---|
| S-(−)-verapamil | f | 1.24 | 4.21 ± 0.03 | 1.95 ± 0.04 |
| R-(+)-verapamil | 2.82 | 1.75 | 5.22 ± 0.08 | 2.29 ± 0.07 |
| Propranolol (IS) | 8.68 | f | 9.13 ± 0.04 | 3.38 ± 0.04 |
| Parameters | S-(−)-Verapamil | R-(+)-Verapamil |
|---|---|---|
| Concentration range (ng/mL) | 1–450 | 1–450 |
| Intercept (a) | 1.21 × 10−2 | 1.91 × 10−2 |
| Slope (b) | 1.45 × 10−3 | 1.81 × 10−3 |
| Coefficient of determination (r2) | 0.998 | 0.997 |
| S Y/N a | 7.39 × 10−3 | 6.88 × 10−3 |
| Sa b | 4.52 × 10−3 | 3.29 × 10−3 |
| Sb c | 2.74 × 10−3 | 5.14 × 10−3 |
| LLOQ (ng/mL) | 1.0 | 1.0 |
| LLOD (ng/mL) | 0.3 | 0.3 |
| Analyte | Intra-Day | Inter-Day | ||||||
|---|---|---|---|---|---|---|---|---|
| Nominal Conc. (ng/mL) | Mean (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | Mean (ng/mL) | Precision (RSD, %) | Accuracy (RE, %) | ||
| S-(−)- verapamil | LLOQ | 1 | 1.03 ± 0.10 | 9.72 | 3.00 | 1.04 ± 0.08 | 7.69 | 4.00 |
| QCL | 3 | 3.14 ± 0.33 | 10.41 | 4.66 | 3.16 ± 0.36 | 11.61 | 5.33 | |
| QCM | 200 | 197.00 ± 7.3 | 3.82 | −1.50 | 198.00 ± 7.54 | 3.81 | −1.00 | |
| QCH | 400 | 390.00 ± 8.55 | 2.19 | −2.50 | 387.08 ± 12.04 | 3.11 | −3.23 | |
| R-(+)- verapamil | LLOQ | 1 | 1.04 ± 0.11 | 10.19 | 4.00 | 1.05 ± 0.09 | 8.14 | 5.00 |
| QCL | 3 | 3.11 ± 0.28 | 9.15 | 3.66 | 3.09 ± 0.30 | 9.82 | 3.00 | |
| QCM | 200 | 197.22 ± 8.30 | 4.21 | 1.39 | 196.50 ± 7.29 | 3.71 | 1.75 | |
| QCH | 400 | 387.60 ± 11.43 | 2.95 | −3.10 | 390.68 ± 10.97 | 2.81 | −2.33 |
| Nominal Concentration (ng/mL) | S-(−)-Verapamil | R-(+)-Verapamil | IS | ||||
|---|---|---|---|---|---|---|---|
| 3 | 200 | 400 | 3 | 200 | 400 | 500 (ng/mL) | |
| Mean a | 2.77 | 191.50 | 392.88 | 2.76 | 193.70 | 391.64 | 470.55 |
| RSD | 9.51 | 4.22 | 2.71 | 7.19 | 3.82 | 2.83 | 8.91 |
| Recovery (%) | 92.33 | 95.75 | 98.22 | 92.00 | 96.85 | 97.91 | 94.70 |
| Mean recovery (%) | 95.58 | 94.92 | 94.11 | ||||
| Analyte | Concentration (ng/mL) | Short Term Stability at Room Temperature (24 h) | Freeze and Thaw Stability at −80 °C (3 cycles) | Long Term Stability at −80 °C (30 days) | Autosampler Stability at 10 °C (24 h) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| RE (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | RE (%) | RSD (%) | |||
| S-(−)-verapamil | QCL | 3 | −5.11 | 9,70 | −6.22 | 11.40 | −4.82 | 11.31 | −5.21 | 9.29 |
| QCH | 400 | 1.42 | 1.81 | 2.63 | 6.52 | 2.91 | 3.85 | 0.75 | 3.24 | |
| R-(+)-verapamil | QCL | 3 | −4.73 | 10.61 | −7.61 | 11.52 | −5.35 | 5.52 | −3.82 | 5.29 |
| QCH | 400 | 1.29 | 2.76 | 4.34 | 6.35 | 2.52 | 4.45 | 3.73 | 4.44 | |
| N | 3 | 3 | 3 | 3 | ||||||
| Parameter | Unit | S-(−)-VER * | R-(+)-VER * |
|---|---|---|---|
| AUC0-t a | ng/mL·h | 754.10 ± 150.82 | 244.10 ± 48.82 |
| AUC0-∞ b | ng/mL·h | 810.30 ± 162.95 | 262.82 ± 52.51 |
| Cmax c | ng/mL | 97.85 ± 19.62 | 30.25 ± 6.43 |
| Tmax d | h | 2.00 ± 0.41 | 2.00 ± 0.41 |
| Cl/Fe | ng/mL.h | 12.34 ± 2.47 | 38.05 ± 7.61 |
| t1/2 f | h | 6.50 ± 1.30 | 6.50 ± 1.33 |
| MRT0-∞ g | h | 8.97 ± 1.80 | 9.14 ± 1.83 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, M.S.; Hefnawy, M.M.; Al-Majed, A.A.; Alrabiah, H.K.; Algrain, N.A.; Obaidullah, A.J.; Altamimi, A.S.; Bin Jardan, Y.A.; Al-Hossaini, A.M. Development and Validation of a Chiral Liquid Chromatographic Assay for Enantiomeric Separation and Quantification of Verapamil in Rat Plasma: Stereoselective Pharmacokinetic Application. Molecules 2021, 26, 2091. https://doi.org/10.3390/molecules26072091
Mohammed MS, Hefnawy MM, Al-Majed AA, Alrabiah HK, Algrain NA, Obaidullah AJ, Altamimi AS, Bin Jardan YA, Al-Hossaini AM. Development and Validation of a Chiral Liquid Chromatographic Assay for Enantiomeric Separation and Quantification of Verapamil in Rat Plasma: Stereoselective Pharmacokinetic Application. Molecules. 2021; 26(7):2091. https://doi.org/10.3390/molecules26072091
Chicago/Turabian StyleMohammed, Mostafa S., Mohamed M. Hefnawy, Abdulrhman A. Al-Majed, Haitham K. Alrabiah, Nasser A. Algrain, Ahmad J. Obaidullah, Abdulmalik S. Altamimi, Yousef A. Bin Jardan, and Abdullah M. Al-Hossaini. 2021. "Development and Validation of a Chiral Liquid Chromatographic Assay for Enantiomeric Separation and Quantification of Verapamil in Rat Plasma: Stereoselective Pharmacokinetic Application" Molecules 26, no. 7: 2091. https://doi.org/10.3390/molecules26072091
APA StyleMohammed, M. S., Hefnawy, M. M., Al-Majed, A. A., Alrabiah, H. K., Algrain, N. A., Obaidullah, A. J., Altamimi, A. S., Bin Jardan, Y. A., & Al-Hossaini, A. M. (2021). Development and Validation of a Chiral Liquid Chromatographic Assay for Enantiomeric Separation and Quantification of Verapamil in Rat Plasma: Stereoselective Pharmacokinetic Application. Molecules, 26(7), 2091. https://doi.org/10.3390/molecules26072091