
A Zinc-Mediated Deprotective Annulation Approach to New Polycyclic Heterocycles


 , , and
, , and 
Abstract
:1. Introduction
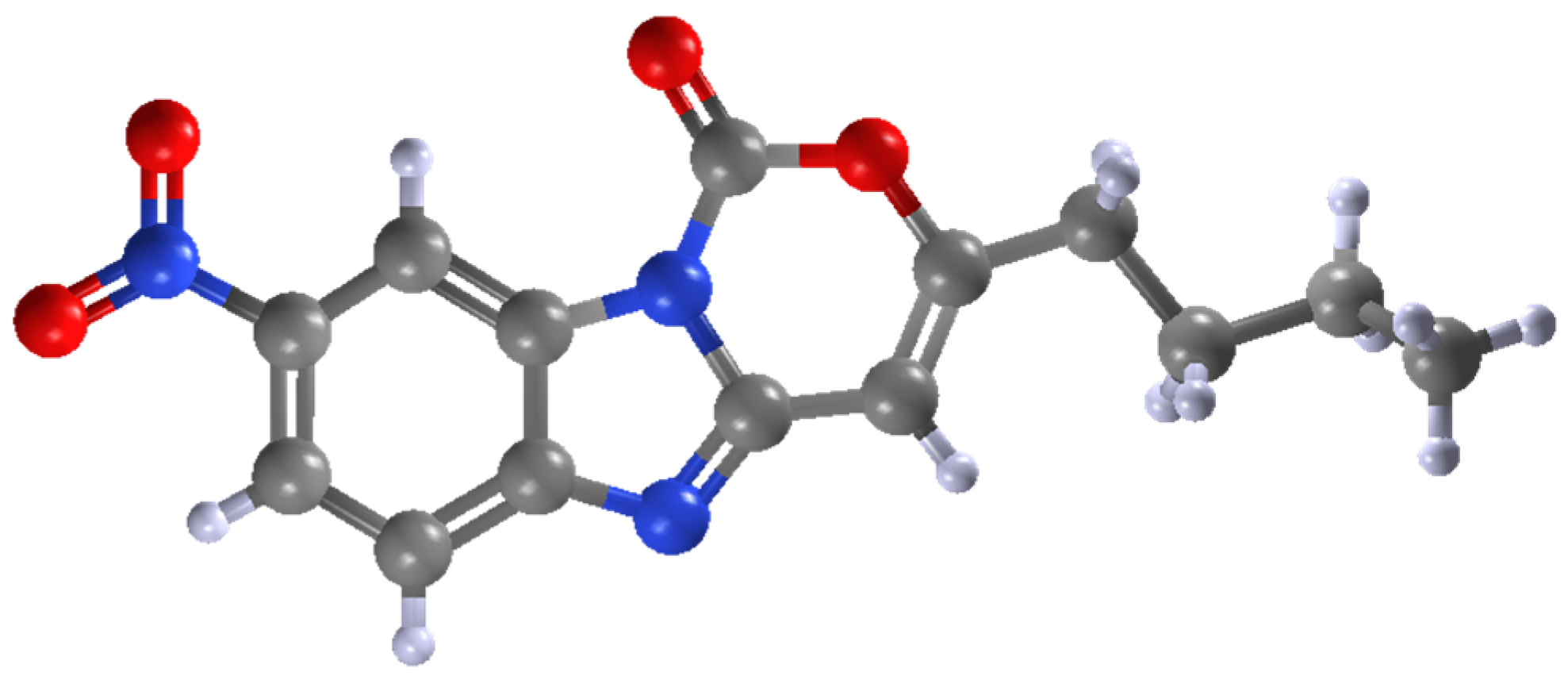
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Methods
3.2. Preparation of Substrates 1
3.3. General Procedure for the Synthesis of Benzimidazoxazinone Derivatives 2
3.3.1. 3-Butyl-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2a
3.3.2. 3-Butyl-7,8-dimethyl-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2b
3.3.3. 3-Butyl-8-methoxy-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2c
3.3.4. 3-Butyl-7-methoxy-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2d
3.3.5. 3-Butyl-7,8-dichloro-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2e
3.3.6. 3-Butyl-8-nitro-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2f
3.3.7. 3-Butyl-7-nitro-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2g
3.3.8. 3-Octyl-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2h
3.3.9. 3-Isopentyl-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2i
3.3.10. 3-Phenethyl-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2j
3.3.11. 3-(Cyclohexylmethyl)-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2k
3.3.12. 3-(Cyclohex-1-en-1-yl)-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2l
3.3.13. 3-(Methoxymethyl)-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2m
3.3.14. Methyl 3-(1-oxo-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-3-yl)propanoate 2n
3.3.15. 3-(2-(tert-Butoxy)ethyl)-1H-benzo[4,5]imidazo[1,2-c][1,3]oxazin-1-one 2o’
3.3.16. 2-(Hex-1-yn-1-yl)-6-nitro-1H-benzo[d]imidazole 3f
3.3.17. 2-(Hex-1-yn-1-yl)-5-nitro-1H-benzo[d]imidazole 3g
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zheng, L.; Hua, R. Recent Advances in Construction of Polycyclic Natural Product Scaffolds via One-Pot Reactions Involving Alkyne Annulation. Front. Chem. 2020, 8, 580355. [Google Scholar] [CrossRef]
- Hong, F.-L.; Ye, L.-W. Transition Metal-Catalyzed Tandem Reactions of Ynamides for Divergent N-Heterocycle Synthesis. Acc. Chem. Res. 2020, 53, 2003–2019. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Verma, Y.; Grewal, P.; Ahlawat, N.; Bhardwaj, P.; Jangid, N.K. Palladium acetate assisted synthesis of five-membered N-polyheterocycles. Synth. Commun. 2020, 50, 1567–1621. [Google Scholar] [CrossRef]
- Gabriele, B.; Mancuso, R.; Veltri, L.; Ziccarelli, I.; Della Ca’, N. Palladium-Catalyzed Double Cyclization Processes Leading to Polycyclic Heterocycles: Recent Advances. Eur. J. Org. Chem. 2019, 2019, 5073–5092. [Google Scholar] [CrossRef]
- Wang, R.; Xie, X.; Liu, H.; Zhou, Y. Rh(III)-Catalyzed C–H Bond Activation for the Construction of Heterocycles with sp3-Carbon Centers. Catalysts 2019, 9, 823. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Carter, R.G. Recent Syntheses and Strategies toward Polycyclic Gelsemium Alkaloids. Angew. Chem. Int. Ed. 2019, 58, 681–694. [Google Scholar] [CrossRef]
- Passador, K.; Thorimbert, S.; Botuha, C. Heteroaromatic Rings of the Future’: Exploration of Unconquered Chemical Space. Synthesis 2019, 51, 384–398. [Google Scholar]
- Hyland, I.K.; O’Toole, R.F.; Smith, J.A.; Bissember, A.C. Progress in the Development of Platelet-Activating Factor Receptor (PAFr) Antagonists and Applications in the Treatment of Inflammatory Diseases. ChemMedChem 2018, 13, 1873–1884. [Google Scholar] [CrossRef]
- Hemmerling, F.; Hahn, F. Biosynthesis of oxygen and nitrogen-containing heterocycles in polyketides. Beilstein J. Org. Chem. 2016, 12, 1512–1550. [Google Scholar] [CrossRef] [Green Version]
- Shokova, E.A.; Kovalev, V.V. Biological Activity of Adamantane-Containing Mono- and Polycyclic Pyrimidine Derivatives (A Review). Pharm. Chem. J. 2016, 50, 63–75. [Google Scholar] [CrossRef]
- Fizer, M.; Slivka, M. Synthesis of [1,2,4]triazolo[1,5-a]pyrimidine (microreview). Chem. Heterocycl. Compds. 2016, 52, 155–157. [Google Scholar] [CrossRef]
- Li, J.; Yang, F.; Hu, W.; Ren, B.; Chen, Z.-S.; Ji, K. Gold(I)-catalyzed tandem cyclization of cyclopropylidene-tethered propargylic alcohols: An approach to functionalized naphtho[2,3-c]pyrans. Chem. Commun. 2020, 56, 9154–9157. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, Z.; Robyens, K.; Van Meervelt, L.; Van der Eycken, R.V. A Gold-Catalyzed Domino Cyclization Enabling Rapid Construction of Diverse Polyheterocyclic Frameworks. Angew. Chem. Int. Ed. 2018, 57, 272–276. [Google Scholar] [CrossRef]
- Alcaide, B.; Almendros, P.; Fernández, I.; Herrea, F.; Luna, A. Gold-Catalyzed Divergent Ring-Closing Modes of Indole-Tethered Amino Allenynes. Chem. Eur. J. 2018, 24, 1448–1454. [Google Scholar] [CrossRef]
- Li, Z.; Song, L.; Van Meervelt, L.; Tian, G.; Van der Eycken, E.V. Cationic Gold(I)-Catalyzed Cascade Bicyclizations for Divergent Synthesis of (Spiro)polyheterocycles. ACS Catal. 2018, 8, 6388–6393. [Google Scholar] [CrossRef]
- Zhang, J.-h.; Wei, Y.; Shi, M. Gold-catalyzed ring enlargement and cycloisomerization of alkynylamide tethered alkylidenecyclopropanes. Org. Chem. Front. 2018, 5, 2980–2985. [Google Scholar] [CrossRef]
- Ito, M.; Kawasaki, R.; Kanyiva, K.S.; Shibata, T. Construction of a Polycyclic Conjugated System Containing a Dibenzazepine Moiety by Cationic Gold(I)-Catalyzed Cycloisomerization. Eur. J. Org. Chem. 2016, 2016, 5234–5237. [Google Scholar] [CrossRef]
- Kumar, R.; Arigela, R.K.; Samala, S.; Kundu, B. Diversity Oriented Synthesis of Indoloazepinobenzimidazole and Benzimidazotriazolobenzodiazepine from N1-Alkyne-1,2-diamines. Chem. Eur. J. 2015, 21, 18828–18833. [Google Scholar] [CrossRef]
- Chen, M.; Sun, N.; Xu, W.; Zhao, J.; Wang, G.; Liu, Y. Gold-Catalyzed Ring Expansion of Alkynyl Heterocycles through 1,2-Migration of an Endocyclic Carbon–Heteroatom Bond. Chem. Eur. J. 2015, 21, 18571–18575. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Bao, M.; Yao, R.; Qiu, L.; Xu, X. Palladium-catalyzed carbene/alkyne metathesis with enynones as carbene precursors: Synthesis of fused polyheterocycles. Chem. Commun. 2018, 54, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Tian, N.; Li, Y.; Jia, C.; Li, X.; Wang, L.; Ciu, X. Construction of Fused Polyheterocycles through Sequential [4 + 2] and [3 + 2] Cycloadditions. Org. Lett. 2017, 19, 1658–1661. [Google Scholar] [CrossRef]
- Kumar, S.; Saunthwal, R.K.; Aggarwal, T.; Kotla, S.K.R.; Verma, A.K. Palladium meets copper: One-pot tandem synthesis of pyrido fused heterocycles via Sonogashira conjoined electrophilic cyclization. Org. Biomol. Chem. 2016, 14, 9063–9071. [Google Scholar] [CrossRef] [PubMed]
- Dethe, D.H.; Boda, R. A Novel Pd-Catalysed Annulation Reaction for the Syntheses of Pyrroloindoles and Pyrroloquinolines. Chem. Eur. J. 2016, 22, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ji, H. Rhodium-Catalyzed [4 + 1] Cyclization via C–H Activation for the Synthesis of Divergent Heterocycles Bearing a Quaternary Carbon. J. Org. Chem. 2018, 83, 4650–4656. [Google Scholar] [CrossRef] [PubMed]
- Youn, S.W.; Yoo, H.J. One-Pot Sequential N-Heterocyclic Carbene/Rhodium(III) Catalysis: Synthesis of Fused Polycyclic Isocoumarins. Adv. Synth. Catal. 2017, 359, 2176–2183. [Google Scholar] [CrossRef]
- Ghorai, D.; Choudhury, J. Rhodium(III)–N-Heterocyclic Carbene-Driven Cascade C–H Activation Catalysis. ACS Catal. 2015, 5, 2692–2696. [Google Scholar] [CrossRef]
- Kozak, J.A.; Dodd, J.M.; Harrison, T.J.; Jardine, K.J.; Patrick, B.O.; Dake, G.R. Enamides and Enesulfonamides as Nucleophiles: Formation of Complex Ring Systems through a Platinum(II)-Catalyzed Addition/Friedel−Crafts Pathway. J. Org. Chem. 2009, 74, 6929–6935. [Google Scholar] [CrossRef]
- Marion, F.; Coulomb, J.; Servais, A.; Courillon, C.; Fensterbank, L.; Malacria, M. Radical cascade cyclizations and platinum(II)-catalyzed cycloisomerizations of ynamides. Tetrahedron 2006, 62, 3856–3871. [Google Scholar] [CrossRef]
- Mamane, V.; Hannen, P.; Fürstner, A. Synthesis of Phenanthrenes and Polycyclic Heteroarenes by Transition-Metal Catalyzed Cycloisomerization Reactions. Chem. Eur. J. 2004, 10, 4556–4575. [Google Scholar] [CrossRef]
- Ghosh, K.; Shankar, M.; Rit, R.K.; Dubey, G.; Bharatam, P.V.; Sahoo, A.K. Sulfoximine-Assisted One-Pot Unsymmetrical Multiple Annulation of Arenes: A Combined Experimental and Computational Study. J. Org. Chem. 2018, 83, 9667–9681. [Google Scholar] [CrossRef]
- Miclo, Y.; Garcia, P.; Evanno, Y.; George, P.; Sevrin, M.; Malacria, M.; Gandon, V.; Aubert, C. Synthesis of Orthogonally Protected Angular Nitrogen Polyheterocycles via CpCo-Catalyzed Pyridine Formation. Synlett 2010, 2010, 314–2318. [Google Scholar]
- Hoshimoto, Y.; Ashida, K.; Sasaoka, Y.; Kumar, R.; Kamikawa, K.; Verdaguer, X.; Riera, A.; Ohashi, M.; Ogoshi, S. Efficient Synthesis of Polycyclic γ-Lactams by Catalytic Carbonylation of Ene-Imines via Nickelacycle Intermediates. Angew. Chem. Int. Ed. 2017, 56, 8206–8210. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Z.-S.; Zhai, T.-Y.; Luo, C.; Zhang, Y.-P.; Chen, Y.-B.; Deng, C.; Liu, R.-S.; Ye, L.-W. Copper-Catalyzed Azide–Ynamide Cyclization to Generate α-Imino Copper Carbenes: Divergent and Enantioselective Access to Polycyclic N-Heterocycles. Angew. Chem. Int. Ed. 2020, 59, 17984–17990. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.-F.; Zhu, X.-P.; Li, D.-Y.; Liu, P.-N. Cu-Catalyzed Cascade Annulation of Alkynols with 2-Azidobenzaldehydes: Access to 6H-Isochromeno[4,3-c]quinolone. J. Org. Chem. 2017, 82, 7032–7039. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.E.; Oniwa, K.; Yamamoto, Y.; Jin, T. N-Methyl Transfer Induced Copper-Mediated Oxidative Diamination of Alkynes. Org. Lett. 2016, 18, 2487–2490. [Google Scholar] [CrossRef] [PubMed]
- Mandadapu, A.K.; Sharma, S.K.; Gupta, S.; Krishna, D.G.V.; Kundu, B. Unprecedented Cu-Catalyzed Coupling of Internal 1,3-Diynes with Azides: One-Pot Tandem Cyclizations Involving 1,3-Dipolar Cycloaddition and Carbocyclization Furnishing Naphthotriazoles. Org. Lett. 2011, 13, 3162–3165. [Google Scholar] [CrossRef] [PubMed]
- Bakholdina, A.; Lukin, A.; Bakulina, O.; Guranova, N.; Krasavin, M. Dual use of propargylamine building blocks in the construction of polyheterocyclic scaffolds. Tetrahedron Lett. 2020, 61, 151970. [Google Scholar] [CrossRef]
- Habert, L.; Sallio, R.; Durandetti, M.; Gosmini, C.; Gillaizeau, I. Zinc Chloride Mediated Synthesis of 3H-Oxazol-2-one and Pyrrolo-oxazin-1-one from Ynamide. Eur. J. Org. Chem. 2019, 2019, 5175–5179. [Google Scholar] [CrossRef]
- Muralidhar, B.; Reddy, S.R. Zn(II) Chloride Promoted Benzannulation Strategy for One-Pot Regioselective Synthesis of 6H-Benzo[c]chromenes. ChemistrySelect 2017, 2, 2539–2543. [Google Scholar] [CrossRef]
- Li, L.; Zhou, B.; Wang, Y.-H.; Shu, C.; Pan, Y.-F.; Lu, X.; Ye, L.-W. Zinc-Catalyzed Alkyne Oxidation/C‒H Functionalization: Highly Site-Selective Synthesis of Versatile Isoquinolones and β-Carbolines. Angew. Chem. Int. Ed. 2015, 54, 8245–8249. [Google Scholar] [CrossRef]
- Kim, H.; Tung, T.T.; Park, S.B. Privileged Substructure-Based Diversity-Oriented Synthesis Pathway for Diverse Pyrimidine-Embedded Polyheterocycles. Org. Lett. 2013, 15, 5814–5817. [Google Scholar] [CrossRef]
- Liu, Y.; Zhen, W.; Dai, W.; Wang, F.; Li, X. Silver(I)-Catalyzed Addition-Cyclization of Alkyne-Functionalized Azomethines. Org. Lett. 2013, 15, 874–877. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nishiyama, M.; Imagawa, H.; Nishizawa, M. Hg(OTf)2-Catalyzed cyclization of alkynyl tert-butylcarbonate leading to cyclic enol carbonate. Tetrahedron Lett. 2006, 47, 8369–8373. [Google Scholar] [CrossRef]
- Krishnan, K.K.; Ujwaldev, S.M.; Saranya, S.; Anilkumar, G.; Beller, M. Recent Advances and Perspectives in the Synthesis of Heterocycles via Zinc Catalysis. Adv. Synth. Catal. 2019, 361, 382–404. [Google Scholar] [CrossRef]
- Saranya, S.; Harry, N.A.; Ujwaldev, S.M.; Anilkumar, G. Recent Advances and Perspectives on the Zinc-Catalyzed Nitroaldol (Henry) Reaction. Asian J. Org. Chem. 2017, 6, 1349–1360. [Google Scholar] [CrossRef]
- Thankachan, A.P.; Asha, S.; Sindhu, K.S.; Anilkumar, G. An overview of Zn-catalyzed enantioselective aldol type C-C bond formation. RSC Adv. 2015, 5, 62179–62193. [Google Scholar] [CrossRef]
- Wu, X.-F. Non-Redox-Metal-Catalyzed Redox Reactions: Zinc Catalysts. Chem. Asian J. 2012, 7, 2502–2509. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H. Zinc-Catalyzed Organic Synthesis: C-C, C-N, C-O Bond Formation Reactions. Adv. Synth. Catal. 2012, 354, 3141–3160. [Google Scholar] [CrossRef]
- Wutts, P.G.M. Greene’s Protective Groups in Organic Synthesis, 5th ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 755–939. [Google Scholar]
- Langille, E.; Bottaro, C.S.; Drouin, A. A novel use of catalytic zinc-hydroxyapatite columns for the selective deprotection of N-tert-butyloxycarbonyl (BOC) protecting group using flow chemistry. J. Flow Chem. 2020, 10, 377–387. [Google Scholar] [CrossRef]
- Vu, H.-D.; Renault, J.; Roisnel, T.; Robert, C.; Jéhan, P.; Gouault, N.; Uriac, P. Reactivity of N-Boc-Protected Amino-Ynones in the Presence of Zinc Chloride: Formation of Acetylenic Cyclic Imines and Their Palladium Complexes. Eur. J. Org. Chem. 2015, 2015, 4868–4875. [Google Scholar] [CrossRef]
- Nigam, S.C.; Mann, A.; Taddei, M.; Wermuth, C.-G. Selective Removal of the Tert-Butoxycarbonyl Group from Secondary Amines: ZnBr2 as the Deprotecting Reagent. Synth. Commun. 1989, 19, 3139–3142. [Google Scholar] [CrossRef]
- Wu, Y.-Q.; Limburg, D.C.; Wilkinson, D.E.; Vaal, M.J.; Hamilton, G.S. A mild deprotection procedure for tert-butyl esters and tert-butyl ethers using ZnBr2 in methylene chloride. Tetrahedron Lett. 2000, 41, 2847–2849. [Google Scholar] [CrossRef]
- Kaul, R.; Brouillette, Y.; Sajjadi, Z.; Hansford, K.A.; Lubell, W.D. Selective tert-Butyl Ester Deprotection in the Presence of Acid Labile Protecting Groups with Use of ZnBr2. J. Org. Chem. 2004, 69, 6131–6133. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | ZnX2 (Equiv) | T (°C) | Solvent | Concentration of 1a b | Conversion of 1a (%) c | Yield of 2a (%) d |
|---|---|---|---|---|---|---|
| 1 | ZnBr2 (1) | 25 | CH2Cl2 | 0.5 | 51 | 25 |
| 2 | ZnBr2 (1) | 25 | MeOH | 0.5 | 3 | 0 |
| 3 | ZnBr2 (1) | 25 | acetone | 0.5 | 12 | Traces |
| 4 | ZnBr2 (0.5) | 25 | CH2Cl2 | 0.5 | 9 | 6 |
| 5 | ZnBr2 (1.5) | 25 | CH2Cl2 | 0.5 | 100 | 72 |
| 6 | ZnBr2 (2) | 25 | CH2Cl2 | 0.5 | 100 | 70 |
| 7 | ZnBr2 (1) | 25 | CH2Cl2 | 1.0 | 62 | 33 |
| 8 | ZnBr2 (1) | 25 | CH2Cl2 | 0.2 | 42 | 10 |
| 9 | ZnBr2 (1) | 40 | CH2Cl2 | 0.5 | 100 | 63 |
| 10 | ZnBr2 (1.5) | 40 | CH2Cl2 | 1.0 | 100 | 79 |
| 11 | ZnCl2 (1.5) | 40 | CH2Cl2 | 1.0 | 100 | 82 |
| 12 | ZnI2 (1.5) | 40 | CH2Cl2 | 1.0 | 100 | 77 |

| Entry | 1 | 2 | Yield of 2 (%) b |
|---|---|---|---|
| 1 |  |  | 82 |
| 2 |  |  | 77 |
| 3 |  |  | 76 |
| 4 |  |  | 83 |
| 5 |  |  | 77 |
| 6 |  |  | 45 c |
| 7 |  |  | 30 d |
| 8 |  |  | 85 |
| 9 |  |  | 82 |
| 10 |  |  | 80 |
| 11 |  |  | 70 |
| 12 |  |  | 66 |
| 13 |  |  | 60 |
| 14 |  |  | 74 |
| 15 |  |  | 66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veltri, L.; Amuso, R.; Petrilli, M.; Cuocci, C.; Chiacchio, M.A.; Vitale, P.; Gabriele, B. A Zinc-Mediated Deprotective Annulation Approach to New Polycyclic Heterocycles. Molecules 2021, 26, 2318. https://doi.org/10.3390/molecules26082318
Veltri L, Amuso R, Petrilli M, Cuocci C, Chiacchio MA, Vitale P, Gabriele B. A Zinc-Mediated Deprotective Annulation Approach to New Polycyclic Heterocycles. Molecules. 2021; 26(8):2318. https://doi.org/10.3390/molecules26082318
Chicago/Turabian StyleVeltri, Lucia, Roberta Amuso, Marzia Petrilli, Corrado Cuocci, Maria A. Chiacchio, Paola Vitale, and Bartolo Gabriele. 2021. "A Zinc-Mediated Deprotective Annulation Approach to New Polycyclic Heterocycles" Molecules 26, no. 8: 2318. https://doi.org/10.3390/molecules26082318
APA StyleVeltri, L., Amuso, R., Petrilli, M., Cuocci, C., Chiacchio, M. A., Vitale, P., & Gabriele, B. (2021). A Zinc-Mediated Deprotective Annulation Approach to New Polycyclic Heterocycles. Molecules, 26(8), 2318. https://doi.org/10.3390/molecules26082318