
Dendrimers Functionalized with Palladium Complexes of N-, N,N-, and N,N,N-Ligands




Abstract
:1. Introduction
2. Results
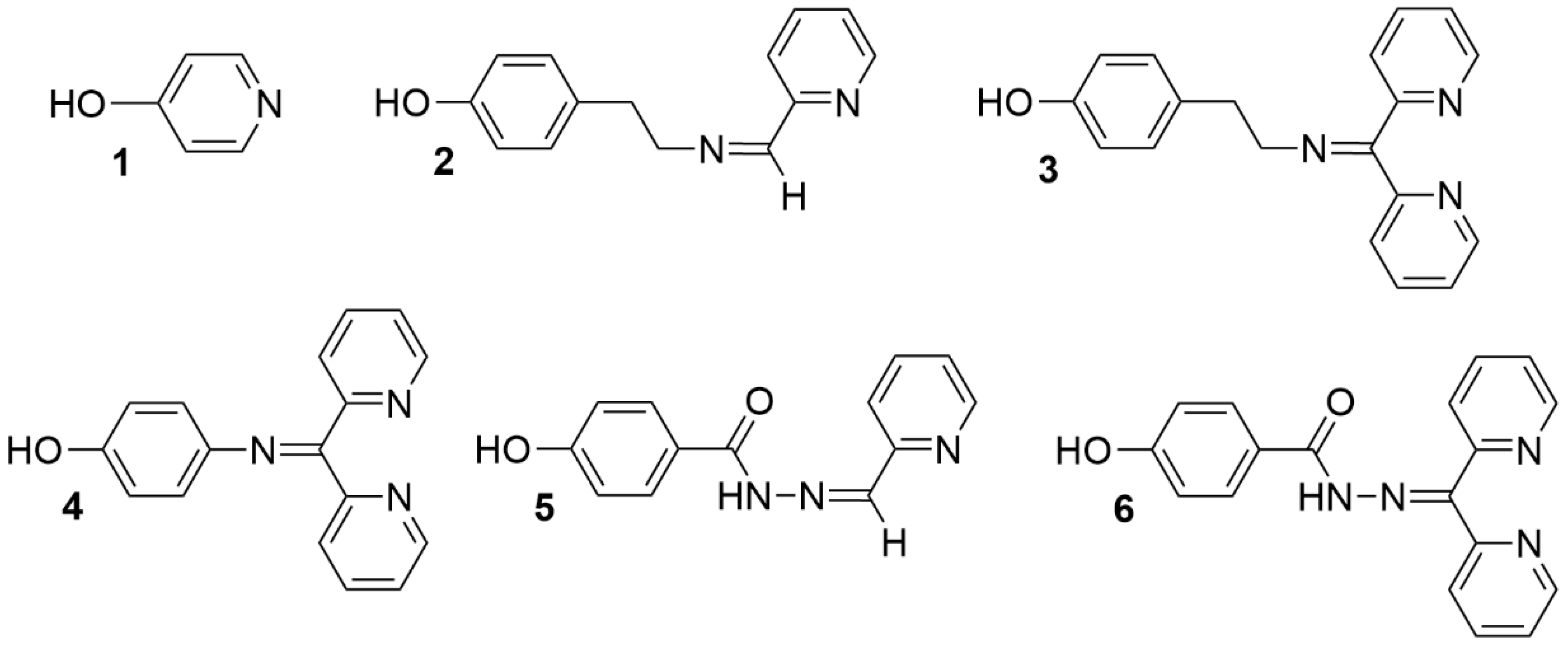
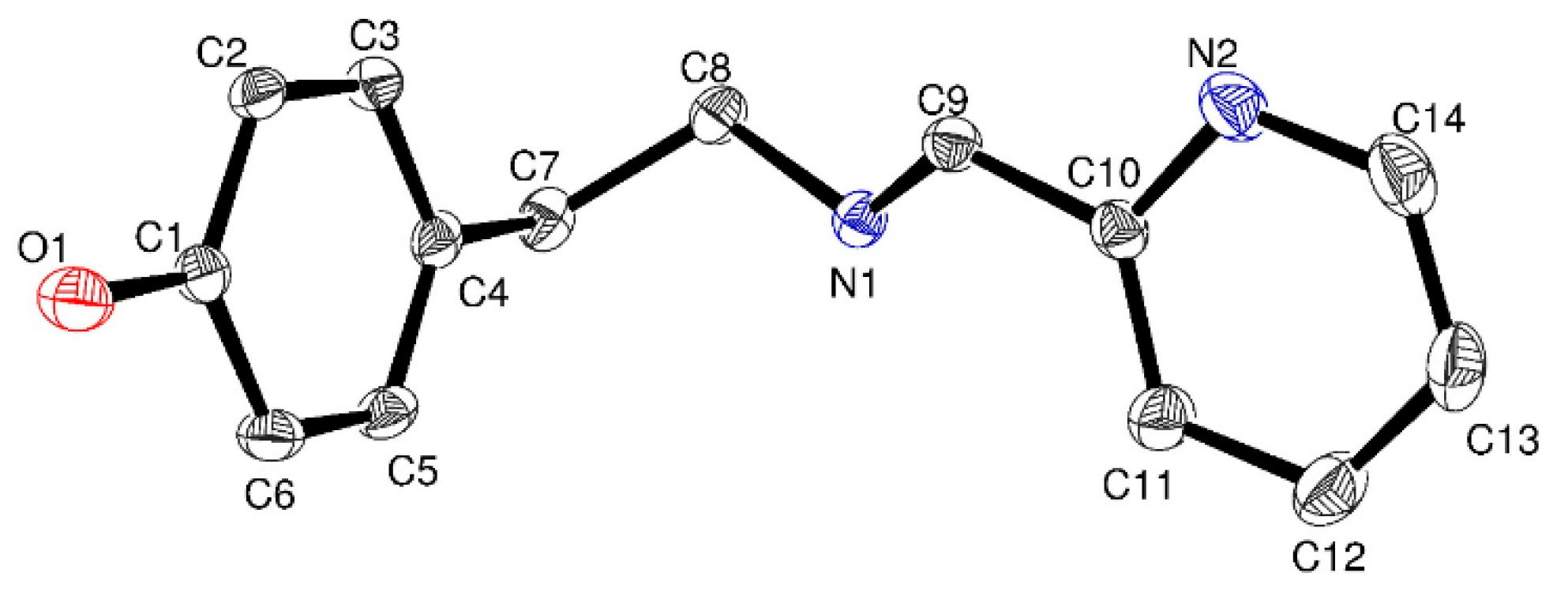
2.1. Synthesis and Characterization of Functionalized Ligands
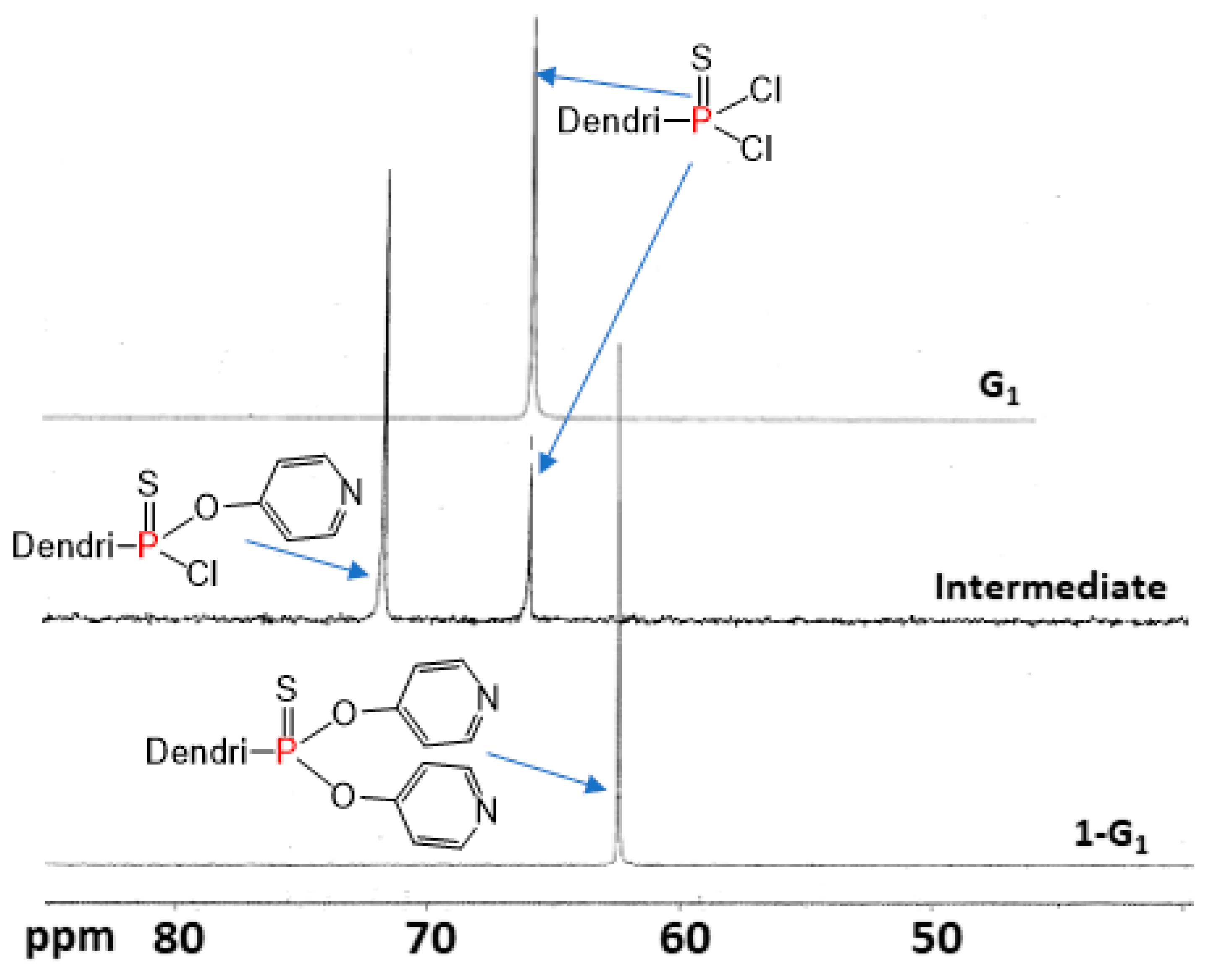
2.2. Grafting of The Functionalized Ligands to The Surface of Dendrimers
2.3. Complexation of Palladium by Monomers and Dendrimers
2.4. Catalytic Attempts
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Astruc, D.; Boisselier, E.; Ornelas, C. Dendrimers Designed for Functions: From Physical, Photophysical, and Supramolecular Properties to Applications in Sensing, Catalysis, Molecular Electronics, Photonics, and Nanomedicine. Chem. Rev. 2010, 110, 1857–1959. [Google Scholar] [CrossRef] [PubMed]
- Caminade, A.M. Inorganic dendrimers. Recent advances for catalysis, nanomaterials, and nanomedicine. Chem. Soc. Rev. 2016, 45, 5174–5186. [Google Scholar] [CrossRef]
- Caminade, A.M.; Fruchon, S.; Turrin, C.O.; Poupot, M.; Ouali, A.; Maraval, A.; Garzoni, M.; Maly, M.; Furer, V.; Kovalenko, V.; et al. The key role of the scaffold on the efficiency of dendrimer nanodrugs. Nat. Comm. 2015, 6, 7722. [Google Scholar] [CrossRef] [PubMed]
- Frey, H.; Schlenk, C. Silicon-based dendrimers. Top. Curr. Chem. 2000, 210(Dendrimers II), 69–129. [Google Scholar]
- Majoral, J.P.; Caminade, A.M.; Maraval, V. The specific contribution of phosphorus in dendrimer chemistry. Chem. Commun. 2002, 2929–2942. [Google Scholar] [CrossRef]
- Galliot, C.; Larré, C.; Caminade, A.M.; Majoral, J.P. Regioselective stepwise growth of dendrimer units in the internal voids of a main dendrimer. Science 1997, 277, 1981–1984. [Google Scholar] [CrossRef]
- Prévôté, D.; Caminade, A.M.; Majoral, J.P. Phosphate, phosphite, ylide and phosphonate terminated dendrimers. J. Org. Chem. 1997, 62, 4834–4841. [Google Scholar] [CrossRef]
- Caminade, A.M.; Ouali, A.; Laurent, R.; Turrin, C.O.; Majoral, J.P. Coordination chemistry with phosphorus dendrimers. Applications as catalysts, for materials, and in biology. Coord. Chem. Rev. 2016, 308, 478–497. [Google Scholar] [CrossRef]
- Ouali, A.; Laurent, R.; Caminade, A.M.; Majoral, J.P.; Taillefer, M. Exaltation of copper catalytic properties in O- and N- arylation and vinylation reactions using phosphorus dendrimers as ligands. J. Am. Chem. Soc. 2006, 128, 15990–15991. [Google Scholar] [CrossRef]
- Keller, M.; Collière, V.; Reiser, O.; Caminade, A.M.; Majoral, J.P.; Ouali, A. Pyrene-tagged dendritic catalysts non-covalently grafted onto magnetic Co/C nanoparticles: An efficient and recyclable system for drug synthesis. Angew. Chem. Int. Ed. 2013, 52, 3626–3629. [Google Scholar] [CrossRef]
- Neumann, P.; Dib, H.; Caminade, A.M.; Hey-Hawkins, E. Redox control of a dendritic homogeneous catalyst. Angew. Chem. Int. Ed. 2015, 54, 311–314. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. The Heck reaction as a sharpening stone of Palladium catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef]
- Chinchilla, R.; Najera, C. Recent advances in Sonogashira reactions. Chem. Soc. Rev. 2011, 40, 5084–5121. [Google Scholar] [CrossRef]
- Mohajer, F.; Heravi, M.M.; Zadsirjan, V.; Poormohammad, N. Copper-free Sonogashira cross-coupling reactions: An overview. RSC Adv. 2021, 11, 6885. [Google Scholar] [CrossRef]
- El Brahmi, N.; El Kazzouli, S.; Mignani, S.M.; Essassi, E.M.; Aubert, G.; Laurent, R.; Caminade, A.M.; Bousmina, M.M.; Cresteil, T.; Majoral, J.P. Original multivalent copper(II)-conjugated phosphorus-dendrimers and corresponding mononuclear copper(II) complexes with anti-tumoral activities. Mol. Pharm. 2013, 10, 1459–1464. [Google Scholar] [CrossRef]
- Karimi, B.; Zamani, A.; Clark, J.H. A bipyridyl palladium complex covalently anchored onto silica as an effective and recoverable interphase catalyst for the aerobic oxidation of alcohols. Organometallics 2005, 24, 4695–4698. [Google Scholar] [CrossRef]
- Karimi, B.; Zamani, A.; Abedi, S.; Clark, J.H. Aerobic oxidation of alcohols using various types of immobilized palladium catalyst: The synergistic role of functionalized ligands, morphology of support, and solvent in generating and stabilizing nanoparticles. Green Chem. 2009, 11, 109–119. [Google Scholar] [CrossRef]
- Andrade-Lopez, N.; Alvarado-Rodriguez, J.G.; Gonzalez-Montiel, S.; Rodriguez-Mendez, M.G.; Paez-Hernandez, M.E.; Galan-Vidal, C.A. Cis-Palladium(II) complexes of derivatives of di-(2-pyridyl)methane: Study of the influence of the bridge group in the coordination mode. Polyhedron 2007, 26, 4825–4832. [Google Scholar] [CrossRef]
- Yorke, J.; Dent, C.; Decken, A.; Xia, A. Synthesis, characterization, and applications of novel di-2-pyridyl imine ligands. Inorg. Chem. Comm. 2010, 13, 54–57. [Google Scholar] [CrossRef]
- Andrade-Lopez, N.; Hanna, T.A.; Alvarado-Rodriguez, J.G.; Luqueño-Reyes, A.; Martínez-Ortega, B.A.; Mendoza-Espinosa, D. Five-membered ring chelate complexes of Ni(II), Pd(II) and Pt(II) derived of di-(2-pyridyl)-N-ethylimine. Polyhedron 2010, 29, 2304–2310. [Google Scholar] [CrossRef]
- Albrecht, K.; Higashimura, H.; Yamamoto, K. Synthesis and properties of Nitrogen-introduced phenylazomethine dendrimer. Synth. Comm. 2014, 44, 2239–2247. [Google Scholar] [CrossRef]
- Gupta, K.C.; Sutar, A.K. Catalytic activities of Schiff base transition metal complexes. Coord. Chem. Rev. 2008, 252, 1420–1450. [Google Scholar] [CrossRef]
- Maraval, V.; Laurent, R.; Caminade, A.M.; Majoral, J.P. Phosphorus-containing dendrimers and their transition metal complexes as efficient recoverable multicenters homogeneous catalysts in organic synthesis. Organometallics 2000, 19, 4025–4029. [Google Scholar] [CrossRef]
- Caminade, A.M.; Laurent, R.; Chaudret, B.; Majoral, J.P. Phosphine-terminated dendrimers: Synthesis and complexation properties. Coord. Chem. Rev. 1998, 178–180, 793–821. [Google Scholar] [CrossRef]
- Agrahari, B.; Layek, S.; Anuradha; Ganguly, R.; Pathak, D.D. Synthesis, crystal structures, and application of two new pincer type palladium(II)-Schiff base complexes in C-C cross-coupling reactions. Inorg. Chim. Acta 2018, 471, 345–354. [Google Scholar] [CrossRef]
- Kumar Rao, G.; Kumar, A.; Pratap Singh, M.; Kumar, A.; Manikrao Biradar, A.K.; Singh, A.K. Influence of pendent alkyl chains on Heck and Sonogashira C-C coupling catalyzed with palladium(II) complexes of selenated Schiff bases having liquid crystalline properties. J. Organomet. Chem. 2014, 753, 42–47. [Google Scholar] [CrossRef]
- Das, P.; Linert, W. Schiff base-derived homogeneous and heterogeneous palladium catalysts for the Suzuki–Miyaura reaction. Coord. Chem. Rev. 2016, 311, 1–23. [Google Scholar] [CrossRef]
- Marmillon, C.; Gauffre, F.; Gulik-Krzywicki, T.; Loup, C.; Caminade, A.M.; Majoral, J.P.; Vors, J.P.; Rump, E. Organophosphorus dendrimers as new gelators for hydrogels. Angew. Chem. Int. Ed. 2001, 40, 2626–2629. [Google Scholar] [CrossRef]
- El Ghzaoui, A.; Gauffre, F.; Caminade, A.M.; Majoral, J.P.; Lannibois-Drean, H. Self-assembly of water-soluble dendrimers into thermoreversible hydrogels and macroscopic fibers. Langmuir 2004, 20, 9348–9353. [Google Scholar] [CrossRef]
- Larpent, C.; Geniès, C.; De Sousa Delgado, A.P.; Caminade, A.M.; Majoral, J.P.; Sassi, J.F.; Leising, F. Giant Dendrimer-Like Particles from Nanolatexes. Chem. Commun 2004, 1816–1817. [Google Scholar] [CrossRef] [PubMed]
- Apartsin, E.K.; Grigoryeva, A.E.; Malrin-Fournol, A.; Ryabchikova, E.I.; Venyaminova, A.G.; Mignani, S.; Caminade, A.M.; Majoral, J.P. Hydrogels of phosphorus dendrimers for biomedical applications: Gelation studies and nucleic acid loading. Pharmaceutics 2018, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Rakowski DuBois, M.; DuBois, D.L. The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation. Chem. Soc. Rev. 2009, 38, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Bian, J.H.; Tong, W.Y.; Pitsch, C.E.; Wu, Y.B.; Wang, X. Mechanism of nickel-catalyzed direct carbonyl-Heck coupling reaction: The crucial role of second-sphere interactions. Dalton Trans. 2021, in press. [Google Scholar] [CrossRef]
- Mandal, P.; Lin, C.H.; Brandão, P.; Mal, D.; Felix, V.; Pratihar, J.L. Synthesis, characterization, structure and catalytic activity of (NNN) tridentate azo-imine nickel(II), palladium(II) and platinum(II) complexes. Polyhedron 2016, 106, 171–177. [Google Scholar] [CrossRef]
- Niknam, E.; Moaddeli, A.; Khalafi-Nezhad, A. Palladium anchored on guanidine-terminated magnetic dendrimer (G3-Gu-Pd): An efficient nano-sized catalyst for phosphorous-free Mizoroki-Heck and copper-free Sonogashira couplings in water. J. Organomet. Chem. 2020, 923, 121369. [Google Scholar] [CrossRef]
- Guo, M.-P.; Liu, S.-W.; Chen, S.-B.; Wen, Y.-J.; Liang, H.; Lv, M.-Y. A Simple and Efficient Palladium Catalyst of Nitrogen-Based Ligand for Cu(I)- and Amine-Free Sonogashira Reaction. Synthetic Comm. 2015, 45, 767–777. [Google Scholar] [CrossRef]
- Alajarin, M.; Lopez-Leonardo, C.; Llamas-Lorente, P.; Raja, R.; Bautista, D.; Orenesa, R.A. Palladium complexes derived from N,N-bidentate NH-iminophosphorane ligands: Synthesis and use as catalysts in the Sonogashira reaction. Dalton Trans. 2012, 41, 12259. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT – Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.C.; Polidori, G.; Camalli, M. SIR92 – a program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- van der Sluis, P.; Spek, A.L. BYPASS – An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. Sect. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Carriedo, G.A.; Gomes Elipe, P.; Garcia Alonso, F.J.; Fernandez-Catuxo, L.; Diaz, M.R.; Garcia Granda, S. Synthesis, X-ray structure and coordination to Mn(CO)3(bipy)+ of the cyclotriphosphazenes N3P3(OC5H4N-2)6 and N3P3(OC5H4N-4)6. J. Organomet. Chem. 1995, 498, 207–212. [Google Scholar] [CrossRef]
- Blais, J.C.; Turrin, C.O.; Caminade, A.M.; Majoral, J.P. MALDI TOF mass spectrometry for the characterization of phosphorus-containing dendrimers. Scope and limitations. Anal. Chem. 2000, 72, 5097–5105. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | T °C | Catalyst Pd | Conversion % |
|---|---|---|---|
| 1 | 40 | 3-Pd | 0 |
| 2 | 40 | 3 + Pd(OAc)2 | 12 |
| 3 | 40 | 3-G1-Pd12 | 0 |
| 4 | 40 | 3-G1 + 12 Pd(OAc)2 | 5 |
| 5 | 70 | 3-Pd | 98 |
| 6 | 70 | 3 + Pd(OAc)2 | 91 |
| 7 | 70 | 3-G1-Pd12 | 90 |
| 8 | 70 | 3-G1 + 12 Pd(OAc)2 | 80 |
| 9 | 70 | 3-G2-Pd24 | 60 |
| 10 | 70 | 3-G3-Pd48 | 66 |
| 11 | 70 | 1-G1 + 12 Pd(OAc)2 | 16 |

| Entry | Mol % Pd | Catalyst Pd | Conversion % |
|---|---|---|---|
| 1 | 1 | 3 + Pd(OAc)2 | 100 |
| 2 | 1 | 3-G1 + 12 Pd(OAc)2 | 100 |
| 3 | 1 | 3-G2 + 24 Pd(OAc)2 | 100 |
| 4 | 1 | 3-G3 + 48 Pd(OAc)2 | 100 |
| 5 | 0.1 | 3 + Pd(OAc)2 | 100 |
| 6 | 0.1 | 3-G1 + 12 Pd(OAc)2 | 100 |
| 7 | 0.1 | 3-G2 + 24 Pd(OAc)2 | 100 |
| 8 | 0.1 | 3-G3 + 48 Pd(OAc)2 | 100 |

| Entry | T °C | Catalyst Pd | Conversion % |
|---|---|---|---|
| 1 | 40 | 2a + Pd(OAc)2 | 64 |
| 2 | 40 | 2-G1 + 12 Pd(OAc)2 | 55 |
| 3 | 40 | 2-G2 + 24 Pd(OAc)2 | 69 |
| 4 | 40 | 2-G3 + 48 Pd(OAc)2 | 71 |
| 5 | 70 | 2a + Pd(OAc)2 | 68 |
| 6 | 70 | 2-G1 + 12 Pd(OAc)2 | 53 |
| 7 | 70 | 2-G2 + 24 Pd(OAc)2 | 61 |
| 8 | 70 | 2-G3 + 48 Pd(OAc)2 | 59 |

| Entry | X | Mol % Pd | Catalyst Pd | Conversion % |
|---|---|---|---|---|
| 1 | I | 1 | 1-G1 + 12 Pd(OAc)2 | 66 |
| 2 | I | 1 | 3 + Pd(OAc)2 | 100 |
| 3 | I | 1 | 3-G1 + 12 Pd(OAc)2 | 100 |
| 4 | I | 1 | 3-G2 + 24 Pd(OAc)2 | 100 |
| 5 | I | 1 | 3-G3 + 48 Pd(OAc)2 | 100 |
| 6 | I | 0.1 | 3 + Pd(OAc)2 | 73 |
| 7 | I | 0.1 | 3-G1 + 12 Pd(OAc)2 | 81 |
| 8 | Br | 1 | 3 + Pd(OAc)2 | 37.5 |
| 9 | Br | 1 | 3-G1 + 12 Pd(OAc)2 | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanbellingen, Q.; Servin, P.; Coinaud, A.; Mallet-Ladeira, S.; Laurent, R.; Caminade, A.-M. Dendrimers Functionalized with Palladium Complexes of N-, N,N-, and N,N,N-Ligands. Molecules 2021, 26, 2333. https://doi.org/10.3390/molecules26082333
Vanbellingen Q, Servin P, Coinaud A, Mallet-Ladeira S, Laurent R, Caminade A-M. Dendrimers Functionalized with Palladium Complexes of N-, N,N-, and N,N,N-Ligands. Molecules. 2021; 26(8):2333. https://doi.org/10.3390/molecules26082333
Chicago/Turabian StyleVanbellingen, Quentin, Paul Servin, Anaïs Coinaud, Sonia Mallet-Ladeira, Régis Laurent, and Anne-Marie Caminade. 2021. "Dendrimers Functionalized with Palladium Complexes of N-, N,N-, and N,N,N-Ligands" Molecules 26, no. 8: 2333. https://doi.org/10.3390/molecules26082333
APA StyleVanbellingen, Q., Servin, P., Coinaud, A., Mallet-Ladeira, S., Laurent, R., & Caminade, A. -M. (2021). Dendrimers Functionalized with Palladium Complexes of N-, N,N-, and N,N,N-Ligands. Molecules, 26(8), 2333. https://doi.org/10.3390/molecules26082333