
Synthesis of New Triazolopyrazine Antimalarial Compounds

 , and
, and
Abstract
:
1. Introduction
2. Results and Discussion
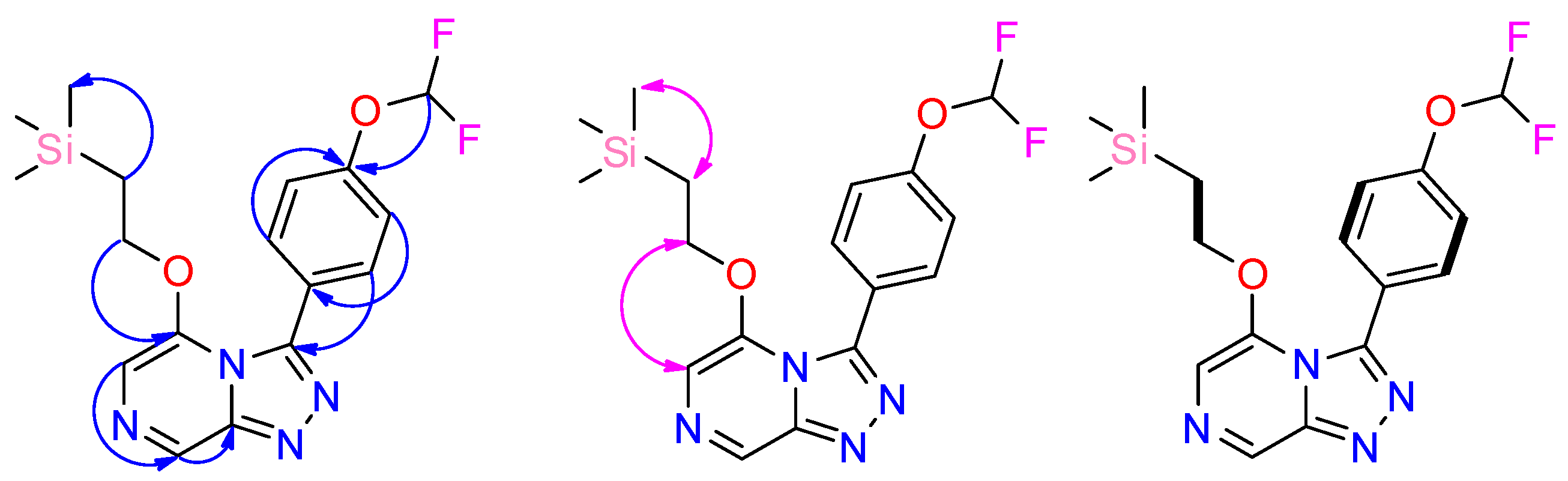
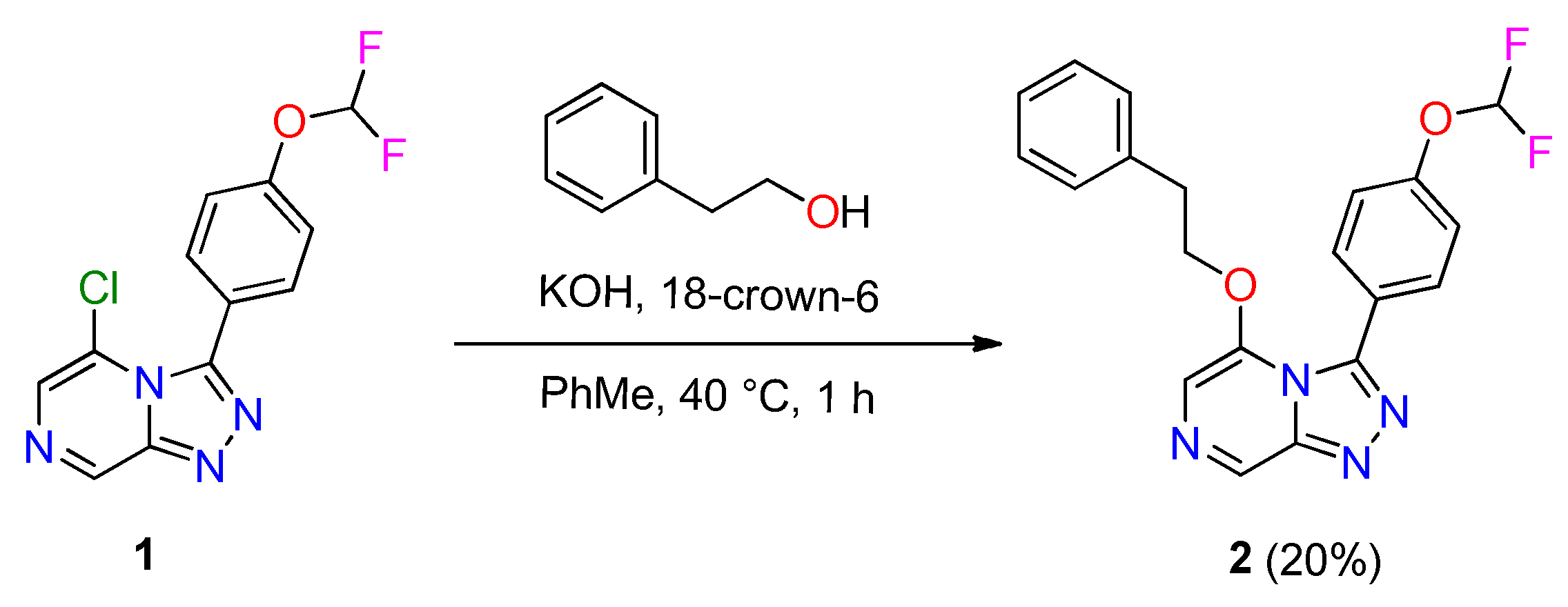
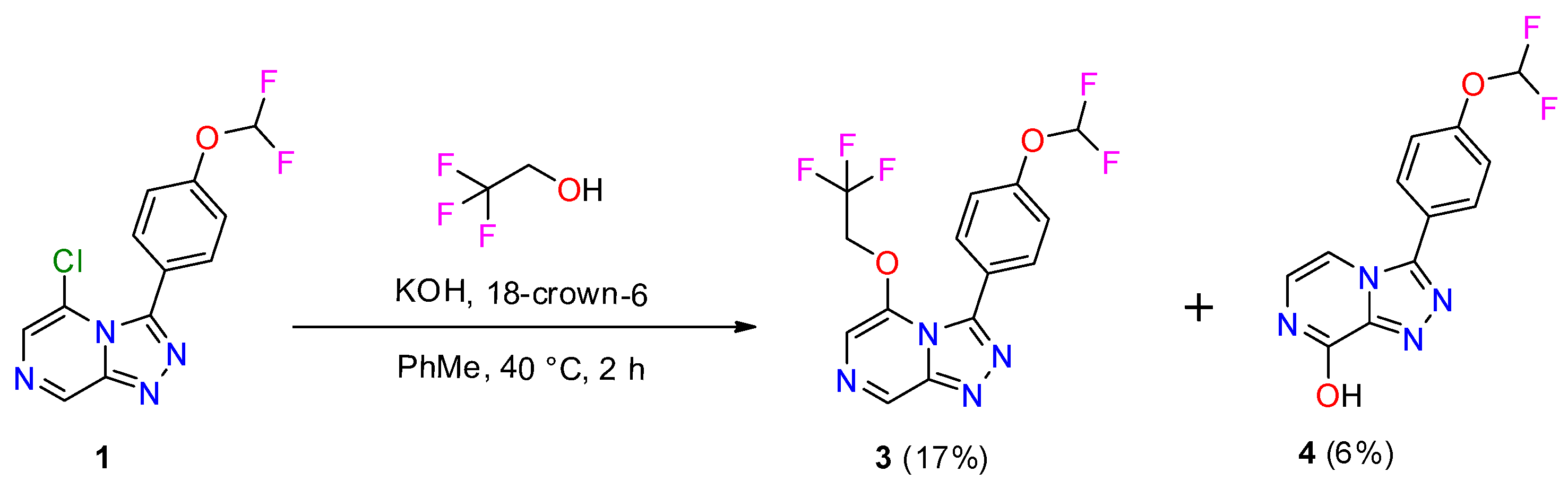
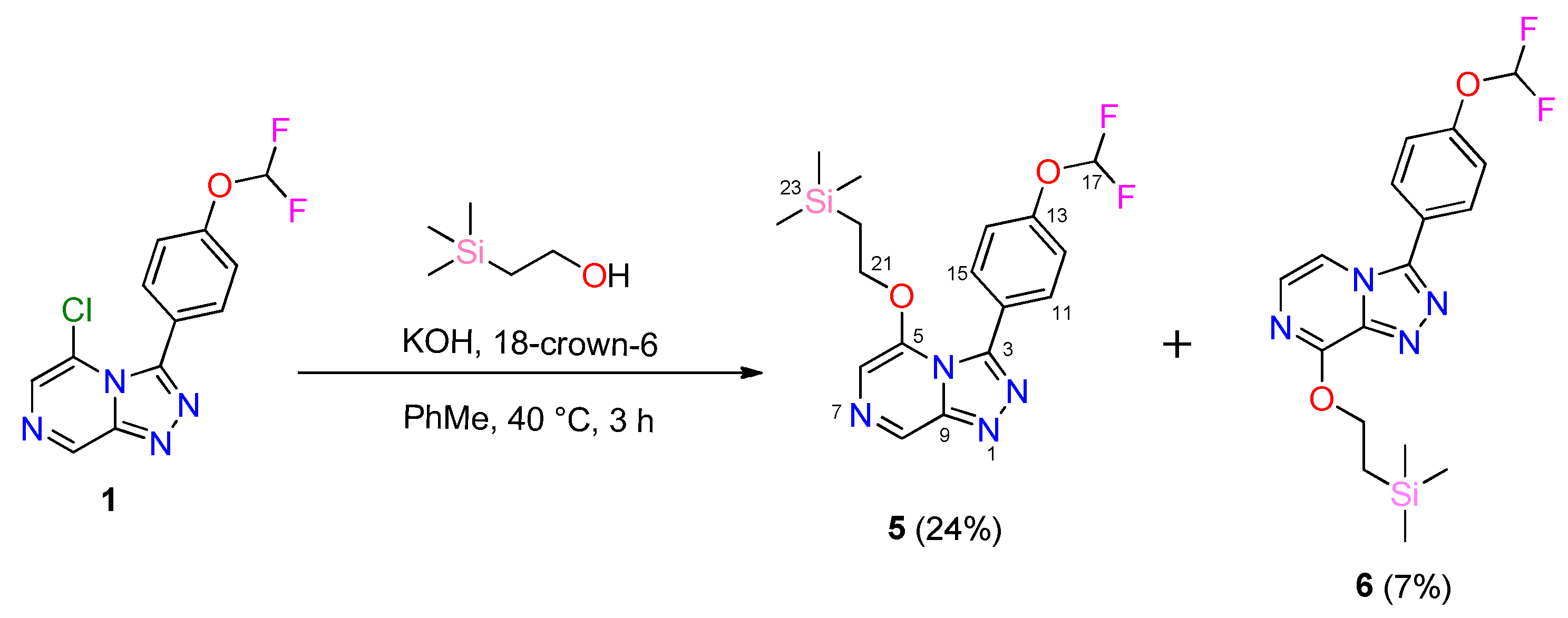
2.1. Synthesis of OSM-Based Scaffolds via Coupling of Primary Alcohols with Chloro-Heterocycles
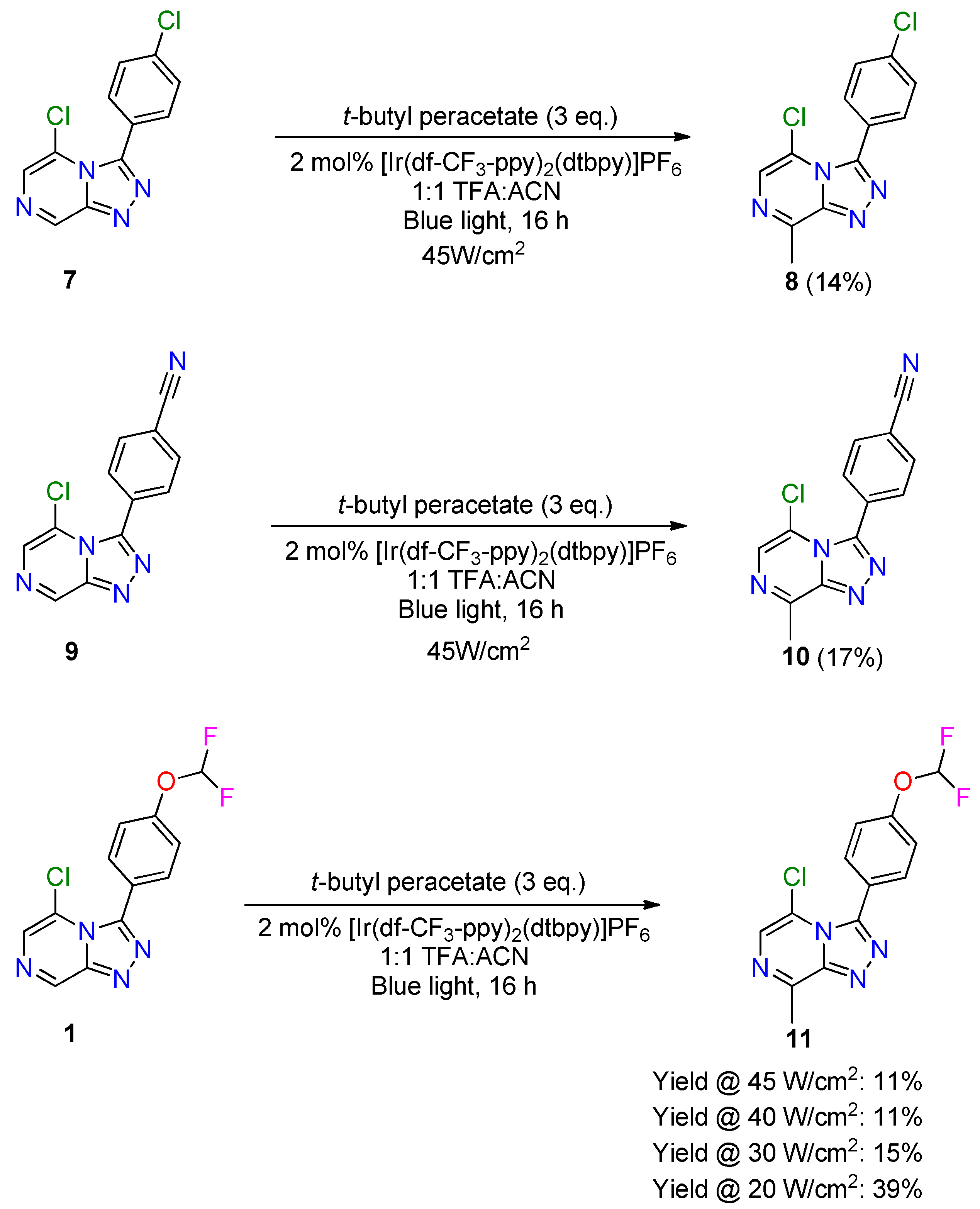
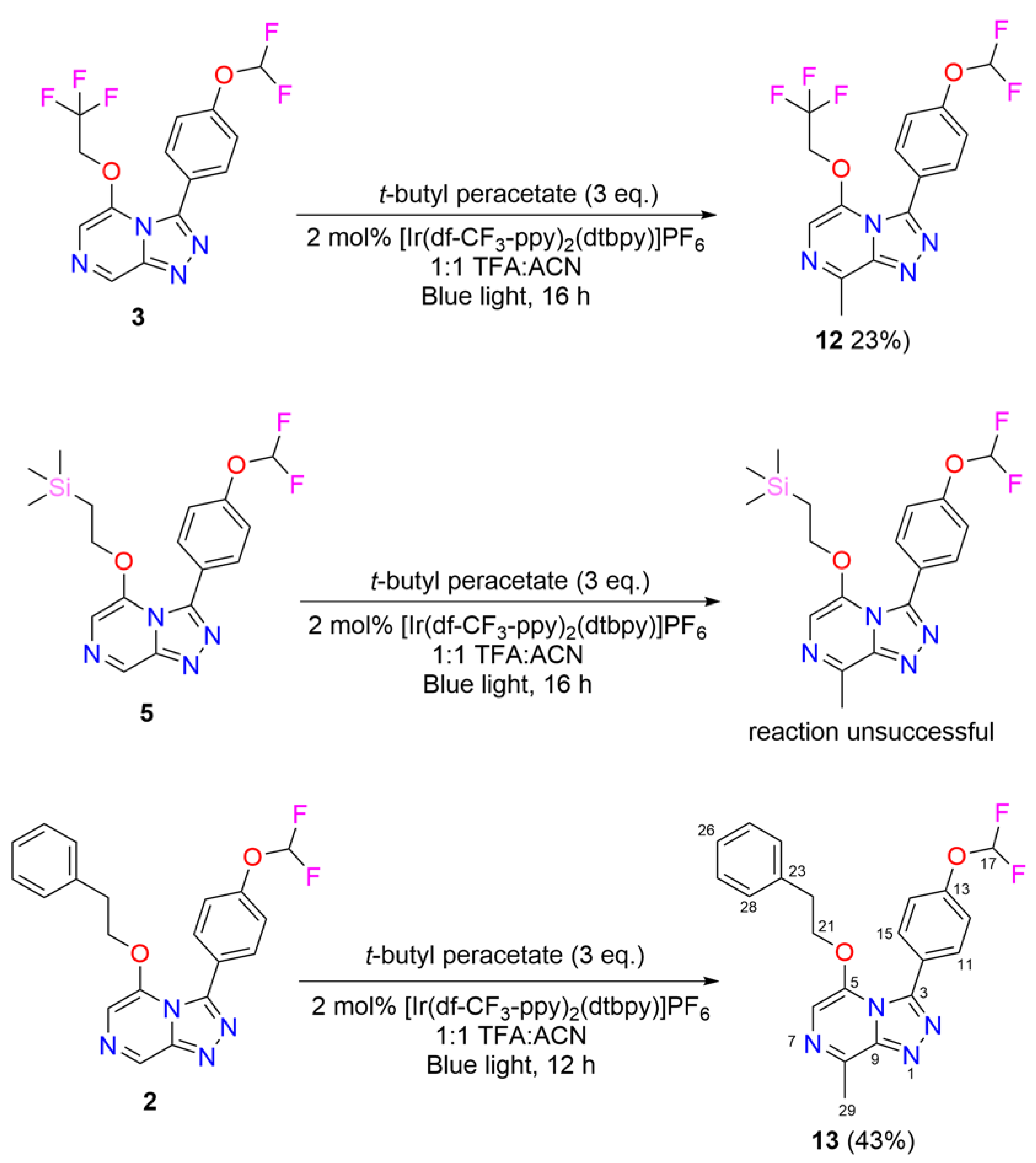
2.2. Late-Stage Functionalization: Methylation of Series 4 Scaffolds and Ether Derivatives via Photoredox Catalysis
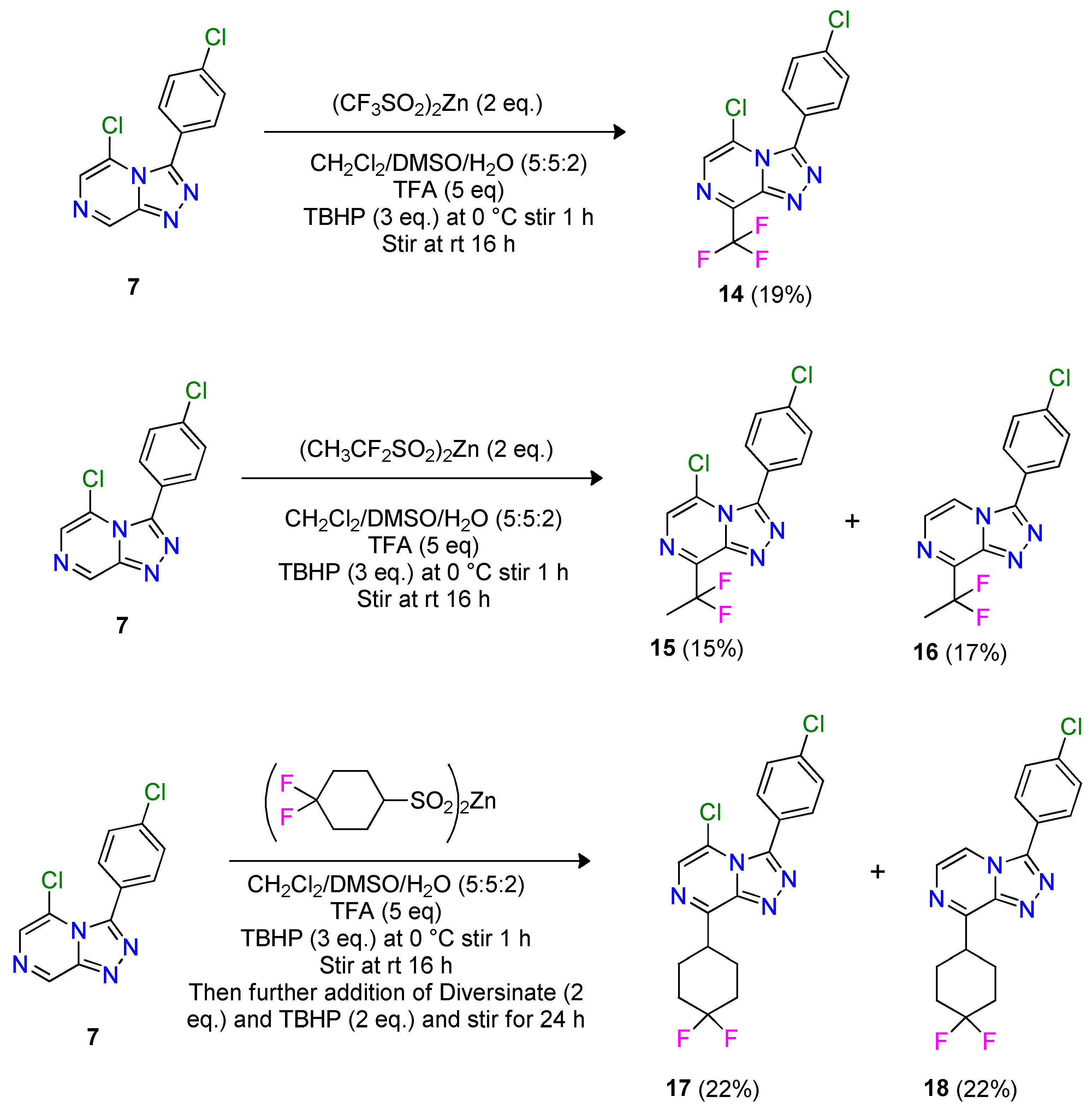
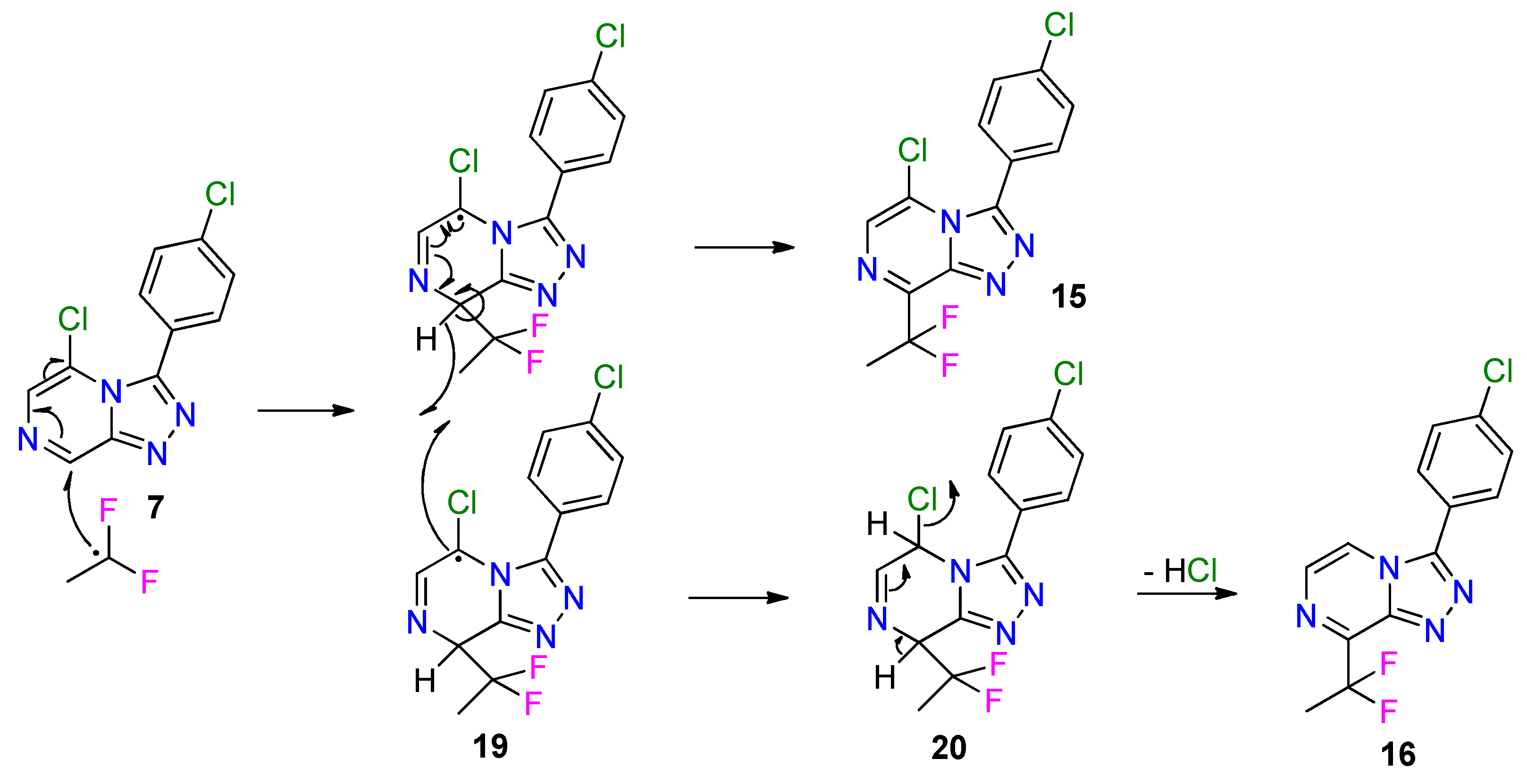
2.3. Late-Stage Functionalization Using Baran Diversinates™
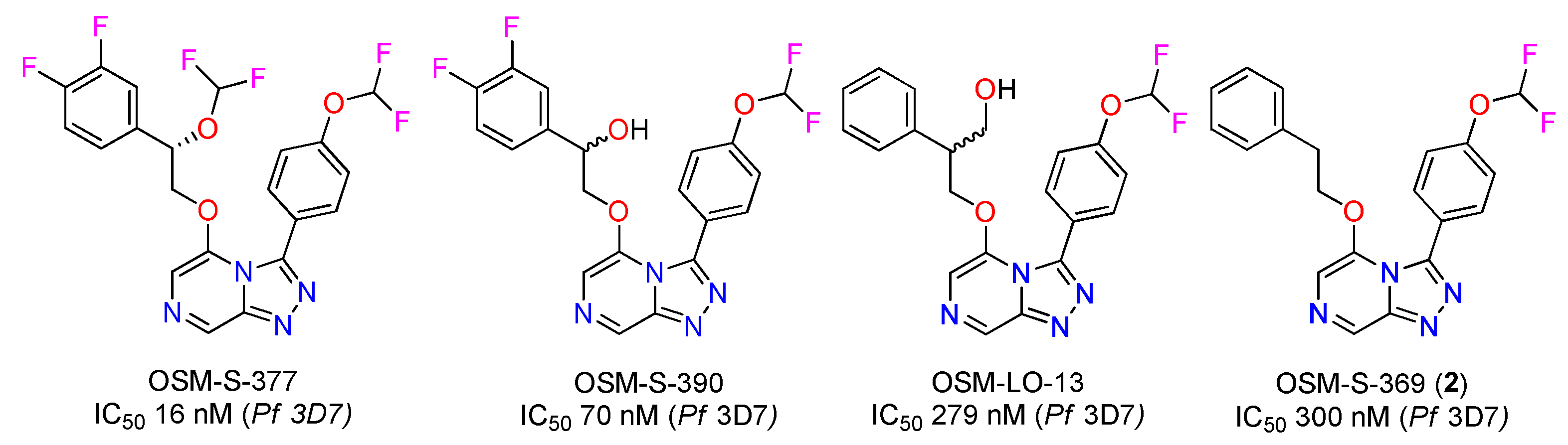
2.4. Bioactivity and Preliminary SAR
3. Materials and Methods
3.1. General Experimental Procedures
3.2. General Procedure for Coupling of Primary Alcohol with Chloro-Heterocycle Scaffold
3.2.1. Compound 2
3.2.2. Compound 3
3.2.3. Compound 4
3.2.4. Compound 5
3.2.5. Compound 6
3.3. General Procedure for Photoredox Catalysis Methylation of Heterocyclic Scaffolds
3.3.1. Compound 8
3.3.2. Compound 10
3.3.3. Compound 11
3.3.4. Compound 12
3.3.5. Compound 13
3.4. General Procedure for the Late-Stage Functionalization Using Baran Diversinates™ on Heterocyclic Scaffolds
3.4.1. Compound 14
3.4.2. Compound 15
3.4.3. Compound 16
3.4.4. Compound 17
3.4.5. Compound 18
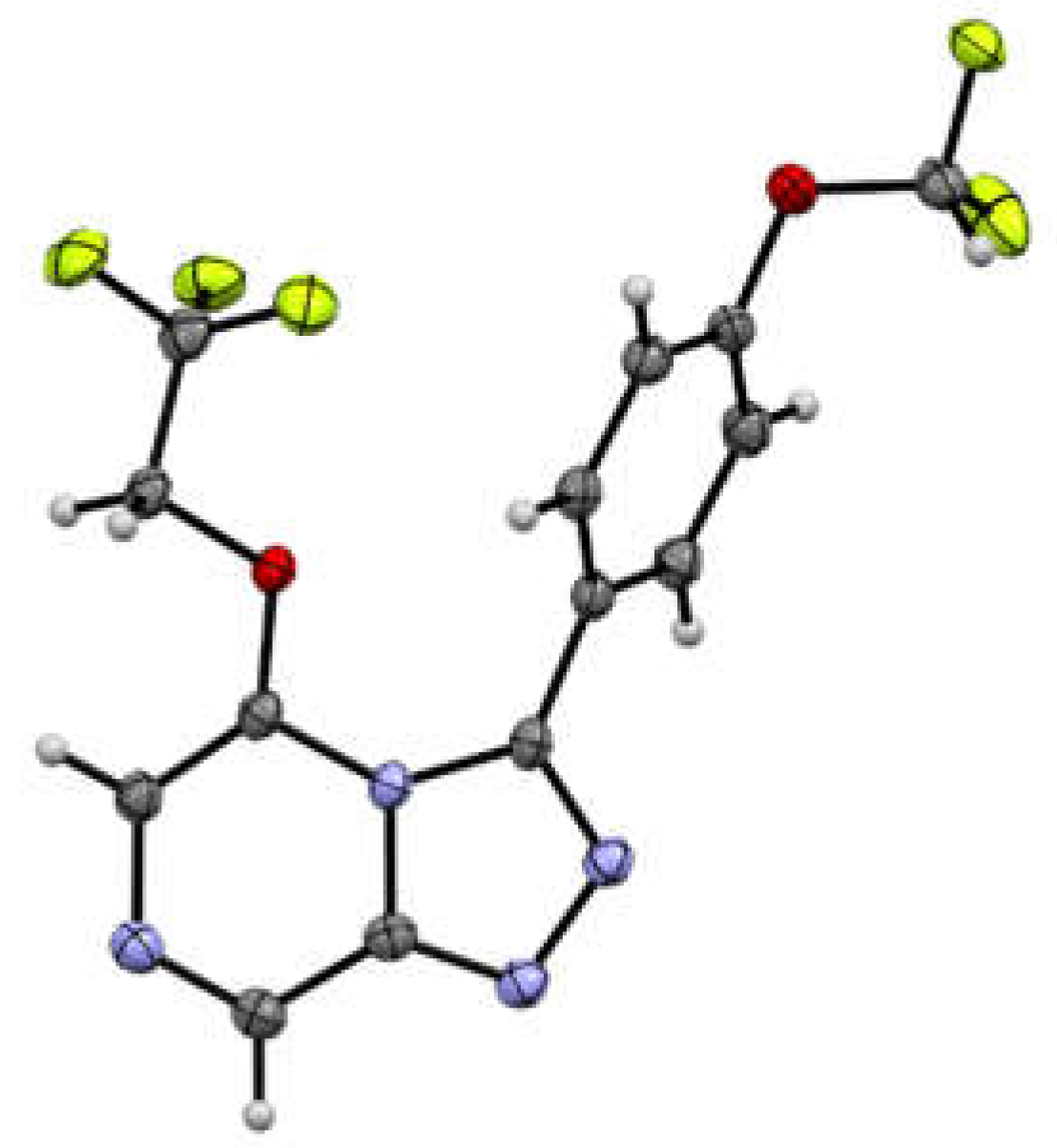
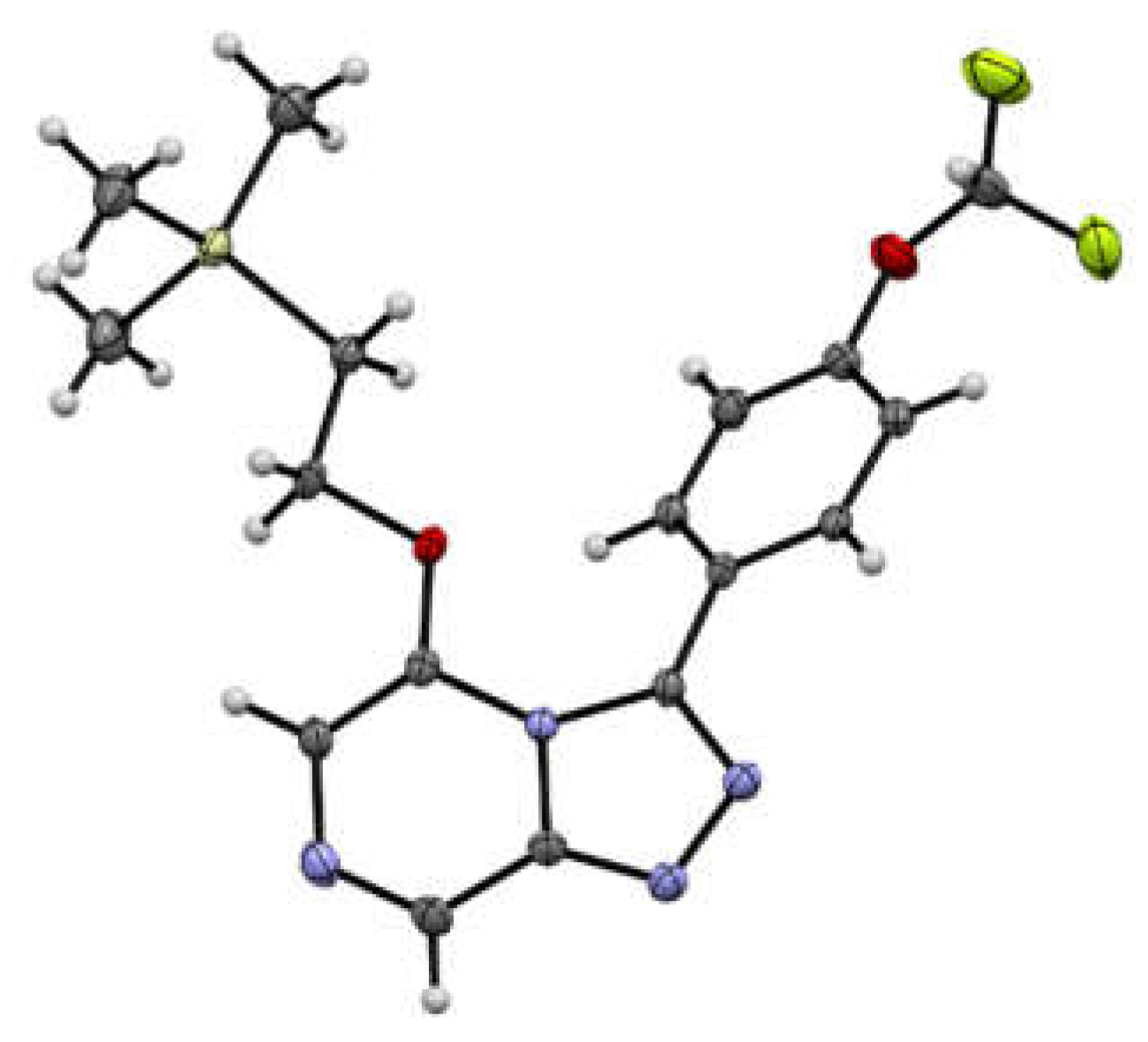
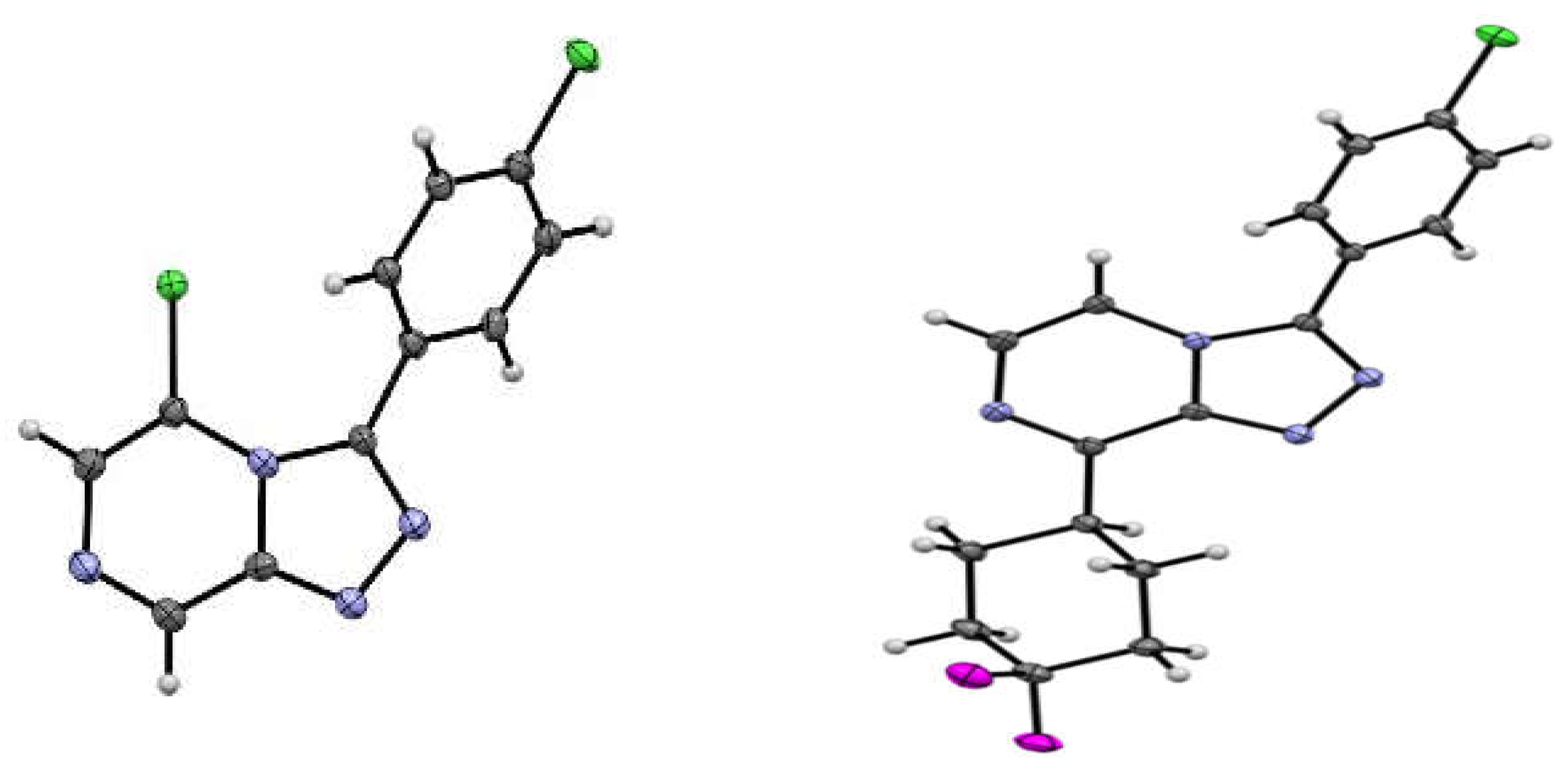
3.5. X-ray Crystallography Studies on Compounds 3, 5, 7, and 18
3.5.1. Crystal Data for Compound 3
3.5.2. Crystal Data for Compound 5
3.5.3. Crystal Data for Compound 7
3.5.4. Crystal Data for Compound 18
3.6. In Vitro Anti-Plasmodial Image-Based Asexual Assay
3.7. In Vitro Cytotoxicity Assay
3.8. Biological Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Shapiro, G. 3,3-Difluoropiperidine Carbamate Heterocyclic Compounds as NR2B NMDA Receptor Antagonists. U.S. Patent 10294230B2, 1 June 2015. [Google Scholar]
- Pasternak, A.; Pio, B.; Chobanian, R.; Harry; Shi, Z.-C.; Dong, S.; Guo, Y.; Walsh, P.; Shawn; Guo, Z. Inhibitors of the renal outer medullary potassium channel. J. Org. Chem. 2020, 85, 13438–13452. [Google Scholar] [CrossRef]
- Lee, S.; Çil, O.; Diez-Cecilia, E.; Anderson, M.O.; Verkman, A.S. Nanomolar-potency 1,2,4-triazoloquinoxaline inhibitors of the kidney urea transporter UT-A1. J. Med. Chem. 2018, 61, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Lee, J.; Go, A.; Choi, G.; Lee, K. Discovery of novel [1,2,4]triazolo[4,3-a]quinoxaline aminophenyl derivatives as BET inhibitors for cancer treatment. Bioorg. Med. Chem. Lett. 2017, 27, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Open Source Malaria Project wiki; Open Source Malaria project/Series 4. Available online: https://github.com/OpenSourceMalaria/Series4/wiki (accessed on 20 January 2021).
- Open Source Malaria Project Wiki; A new triazolopyrazine series for OSM series 4. Available online: http://malaria.ourexperiment.org/osdd_malaria_shared/7949/A_New_Triazolopyrazine_Series_for_OSM__Series_4.html (accessed on 20 January 2021).
- Open Source Malaria Project wiki; Mechanism of Action: Possible PfATP4 Activity Deduced from Parasite Ion Regulation Asays. Available online: https://github.com/OpenSourceMalaria/Series4/wiki/Mechanism-of-Action%3A-Possible-PfATP4-Activity-Deduced-from-Parasite-Ion-Regulation-Assays (accessed on 22 January 2021).
- Gilson, P.R.; Kumarasingha, R.; Thompson, J.; Zhang, X.; Penington, J.S.; Kalhor, R.; Bullen, H.E.; Lehane, A.M.; Dans, M.G.; De Koning-Ward, T.F.; et al. A 4-cyano-3-methylisoquinoline inhibitor of Plasmodium falciparum growth targets the sodium efflux pump PfATP4. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidya, A.B.; Morrisey, J.M.; Zhang, Z.; Das, S.; Daly, T.M.; Otto, T.D.; Spillman, N.J.; Wyvratt, M.; Siegl, P.; Marfurt, J.; et al. Pyrazoleamide compounds are potent antimalarials that target Na+ homeostasis in intraerythrocytic Plasmodium falciparum. Nat. Commun. 2014, 5, 5521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillman, N.J.; Kirk, K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosling, J.E.O.; Ridgway, M.C.; Summers, R.L.; Kirk, K.; Lehane, A.M. Biochemical characterization and chemical inhibition of PfATP4-associated Na+-ATPase activity in Plasmodium falciparum membranes. J. Biol. Chem. 2018, 293, 13327–13337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Open Source Malaria Project Wiki; Synthesis of the Ether Linked Series. Available online: https://github.com/OpenSourceMalaria/Series4/wiki/Synthesis-of-the-Ether-Linked-Series (accessed on 23 January 2021).
- Tse, E. Open Source Malaria: Potent Triazolopyrazine-Based Antiplasmodium Agents That Probe an Important Mechanism of Action. Ph.D. Thesis, University of Sydney, Sydney, Australia, January 2019. [Google Scholar]
- Open Source Malaria Project wiki; Compounds sent for testing in Dundee, December 2017 #10. Available online: https://github.com/OpenSourceMalaria/Series4/issues/10 (accessed on 20 January 2021).
- Open Source Malaria Project wiki; OpenSourceMalaria/Series4/Sources of Data. Available online: https://github.com/OpenSourceMalaria/Series4/wiki/Sources-of-Data (accessed on 20 February 2021).
- Eryilmaz, E. Multi-targeted anti-leukemic drug design with the incorporation of silicon into Nelarabine: How silicon increases bioactivity. Eur. J. Pharm. Sci. 2019, 134, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Korsik, M.; Tse, E.G.; Smith, D.G.; Lewis, W.; Rutledge, P.J.; Todd, M.H. tele-Substitution reactions in the synthesis of a prom-ising class of 1,2,4-triazolo[4,3-a]pyrazine-based antimalarials. J. Org. Chem. 2020, 85, 13438–13452. [Google Scholar] [CrossRef] [PubMed]
- DiRocco, D.A.; Dykstra, K.; Krska, S.; Vachal, P.; Conway, D.V.; Tudge, M. Late-stage functionalization of biologically active heterocycles through photoredox catalysis. Angew. Chem. Int. Ed. 2014, 53, 4802–4806. [Google Scholar] [CrossRef] [PubMed]
- Verschueren, R.H.; De Borggraeve, W.M. Electrochemistry and photoredox catalysis: A comparative evaluation in organic synthesis. Molecules 2019, 24, 2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuttruff, C.A.; Haile, M.; Kraml, J.; Tautermann, C.S. Late-stage functionalization of drug-like molecules using Diversinates. ChemMedChem 2018, 13, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; Von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Duffy, S.; Avery, V.M. Development and optimization of a novel 384-well anti-malarial imaging assay validated for high-throughput screening. Am. J. Trop. Med. Hyg. 2012, 86, 84–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, S.; Avery, V.M. A novel approach for the discovery of chemically diverse anti-malarial compounds targeting the Plasmodium falciparum coenzyme A synthesis pathway. Malar. J. 2014, 13, 343. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. ACTA Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.J.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0– new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
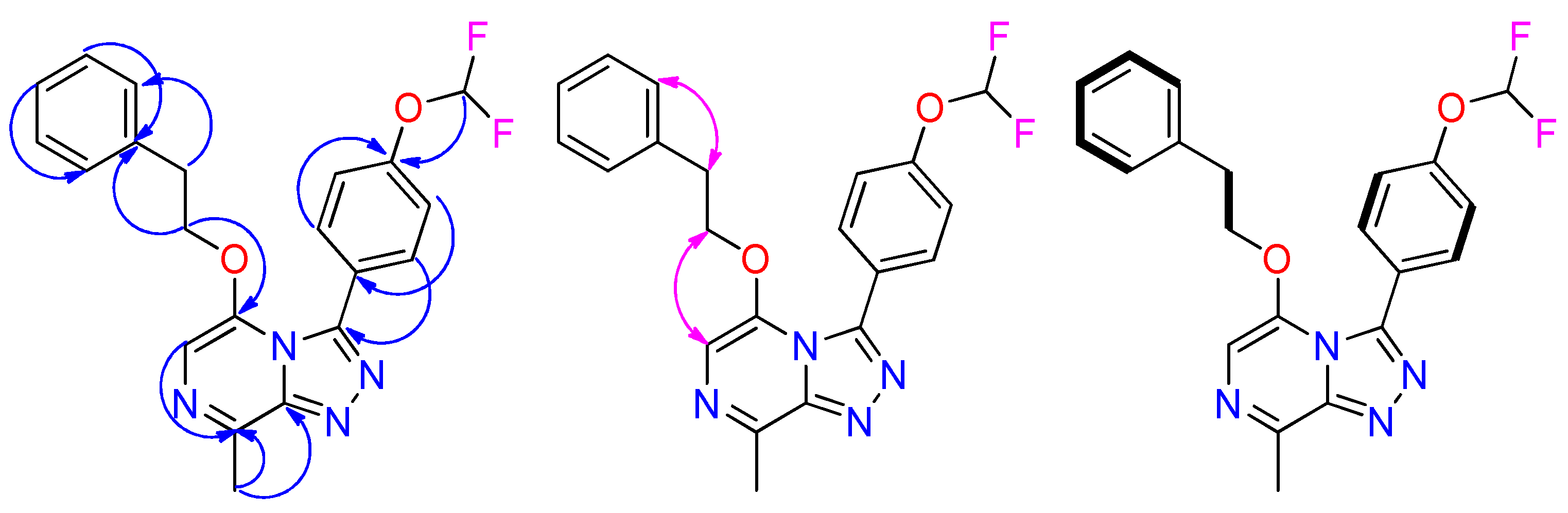
| Position | δH, mult., J in Hz, int. | δC, mult., J in Hz | COSY | HMBC | ROESY |
|---|---|---|---|---|---|
| 3 | 145.6, s | ||||
| 5 | 143.9, s | ||||
| 6 | 7.59, s, 1 H | 109.1, s | 5, 8 | 21 | |
| 8 | 9.01, s, 1 H | 134.7, s | 6, 9 | ||
| 9 | 147.4, s | ||||
| 10 | 124.8, s | ||||
| 11 | 7.81, m, 1 H | 132.8, s | 12 | 3, 13,15 | 12 |
| 12 | 7.31, m, 1 H | 117.6, s | 11 | 10,14 | 11 |
| 13 | 151.9, t, 3.3 | ||||
| 14 | 7.31, m, 1 H | 117.6, s | 15 | 10, 12 | 15 |
| 15 | 7.81, m, 1 H | 132.8, s | 14 | 3, 11, 13 | 14 |
| 17 | 7.36, t, 73.6, 1 H | 116.1, t, 258.5 | 13 | ||
| 21 | 4.33, m, 2 H | 69.4, s | 22 | 5, 22 | 6, 22 |
| 22 | 0.91, m, 2 H | 16.5, s | 21 | 21, 24, 25, 26 | 21, 24, 25, 26 |
| 24 | 0.07, s, 3 H | −1.7, s | 22, 25, 26 | 22 | |
| 25 | 0.07, s, 3 H | −1.7, s | 22, 24, 26 | 22 | |
| 26 | 0.07, s, 3 H | −1.7, s | 22, 24, 25 | 22 |
| Position | 1H, mult., J in Hz, int | 13C, mult., J in Hz | COSY | HMBC | ROESY |
|---|---|---|---|---|---|
| 3 | 146.3, s | ||||
| 5 | 142.8, s | ||||
| 6 | 7.39, s, 1 H | 107.9, s | 5, 8 | ||
| 8 | 143.2, s | ||||
| 9 | 146.8, s | ||||
| 10 | 124.8, s | ||||
| 11 | 7.74, m, 1 H | 132.6, s | 12 | 3, 13,15 | 12 |
| 12 | 7.29, m, 1 H | 117.6, s | 11 | 10,14 | 11 |
| 13 | 152.0, t, 3.2 | ||||
| 14 | 7.29, m, 1 H | 117.6, s | 15 | 10, 12 | 15 |
| 15 | 7.74, m, 1 H | 132.6, s | 14 | 3, 11, 13 | 14 |
| 17 | 7.36, t, 73.7, 1 H | 116.2, t, 258.2 | 13 | ||
| 21 | 4.42, t, 6.5, 2 H | 71.0, s | 22 | 5, 22, 23 | 6, 22 |
| 22 | 2.86, t, 6.5, 2 H | 33.9, s | 21 | 21, 23, 24, 28 | 21, 24, 28 |
| 23 | 137.4, s | ||||
| 24 | 6.90, m, 1 H | 128.7, s | 25 | 22, 26, 28 | 25 |
| 25 26 27 28 29 | 7.17, m, 1 H 7.16, m, 1 H 7.17, m, 1 H 6.90, m, 1 H 2.72, s, 3 H | 128.2, s 126.4, s 128.2, s 128.7, s 19.7, s | 24, 26 25, 27 26, 28 27 | 23, 27 24, 28 23, 25 22, 24, 26 6, 8, 9 | 24, 26 25, 27 26, 28 27 |
| Compounds | Pf 3D7 a IC50 ± SD µM | Pf Dd2 b IC50 ± SD µM | SI for 3D7 c | SI for Dd2 c |
|---|---|---|---|---|
| 1 | 16.81 ± 1.25 d | 22.81 ± 13.73 | >4 | >3 |
| 2 | 0.31 ± 0.01 | 0.54 ± 0.02 | >258 | >148 |
| 3 | 16.65 ± 1.02 | 21.24 ± 5.41 | >4 | >3 |
| 4 | e | NA | - | - |
| 5 | 13.86 ± 0.87 | NA | >5 | - |
| 6 | e | NA | - | - |
| 7 | 12.62 ± 1.90 | 14.32 ± 0.62 | >6 | >5 |
| 8 | 19.18 ± 0.28 d | 32.39 ± 1.44 | >4 | >2 |
| 9 | 18.89 ± 0.66 d | 25.87 ± 0.74 | >4 | >3 |
| 10 | 27.33 ± 0.82 d | 38.25 ± 0.61 | >2 | >2 |
| 11 | 12.49 ± 0.58 | 31.22 ± 0.82 | >6 | >2 |
| 12 | 27.33 ± 0.82 d | e | >2 | |
| 13 | 11.01 ± 0.56 d | 8.20 ± 0.60 | >7 | >9 |
| 14 | 4.84 ± 0.13 | 6.18 ± 0.09 | >16 | >12 |
| 15 | 1.72 ± 0.02 | 2.58 ± 0.07 | >46 | >31 |
| 16 | 6.40 ± 0.10 | 24.70 ± 0.15 | >12 | >3 |
| 17 | 20.19 ± 2.86 d | e | >3 | |
| 18 | e | NA | - | - |
| Controls | Pf 3D7a IC50 ± SD nM | Pf Dd2b IC50 ± SD nM | SI for 3D7c | SI for Dd2c |
| Pyrimethamine | 12.08 ± 0.04 | e | >331 | - |
| Artesunate | 3.48 ± 0.14 | 2.00 ± 0.10 | >2874 | >5000 |
| Puromycin | 95.28 ± 1.45 | 101.00 ± 2.80 | 12 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, D.J.G.; Jenkins, I.D.; Huxley, C.; Coster, M.J.; Lum, K.Y.; White, J.M.; Avery, V.M.; Davis, R.A. Synthesis of New Triazolopyrazine Antimalarial Compounds. Molecules 2021, 26, 2421. https://doi.org/10.3390/molecules26092421
Johnson DJG, Jenkins ID, Huxley C, Coster MJ, Lum KY, White JM, Avery VM, Davis RA. Synthesis of New Triazolopyrazine Antimalarial Compounds. Molecules. 2021; 26(9):2421. https://doi.org/10.3390/molecules26092421
Chicago/Turabian StyleJohnson, Daniel J. G., Ian D. Jenkins, Cohan Huxley, Mark J. Coster, Kah Yean Lum, Jonathan M. White, Vicky M. Avery, and Rohan A. Davis. 2021. "Synthesis of New Triazolopyrazine Antimalarial Compounds" Molecules 26, no. 9: 2421. https://doi.org/10.3390/molecules26092421
APA StyleJohnson, D. J. G., Jenkins, I. D., Huxley, C., Coster, M. J., Lum, K. Y., White, J. M., Avery, V. M., & Davis, R. A. (2021). Synthesis of New Triazolopyrazine Antimalarial Compounds. Molecules, 26(9), 2421. https://doi.org/10.3390/molecules26092421