
Spectral Features of Canthaxanthin in HCP2. A QM/MM Approach



Abstract
:
1. Introduction
2. Results and Discussion
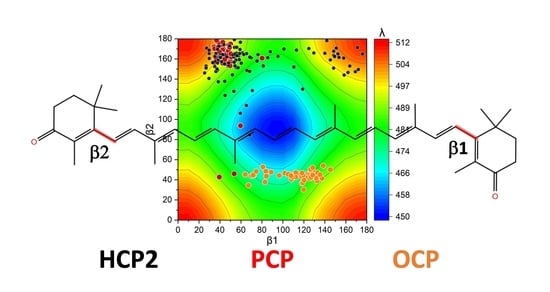
2.1. QM/MM Calculations of HCP2 Models from the Crystal Structure
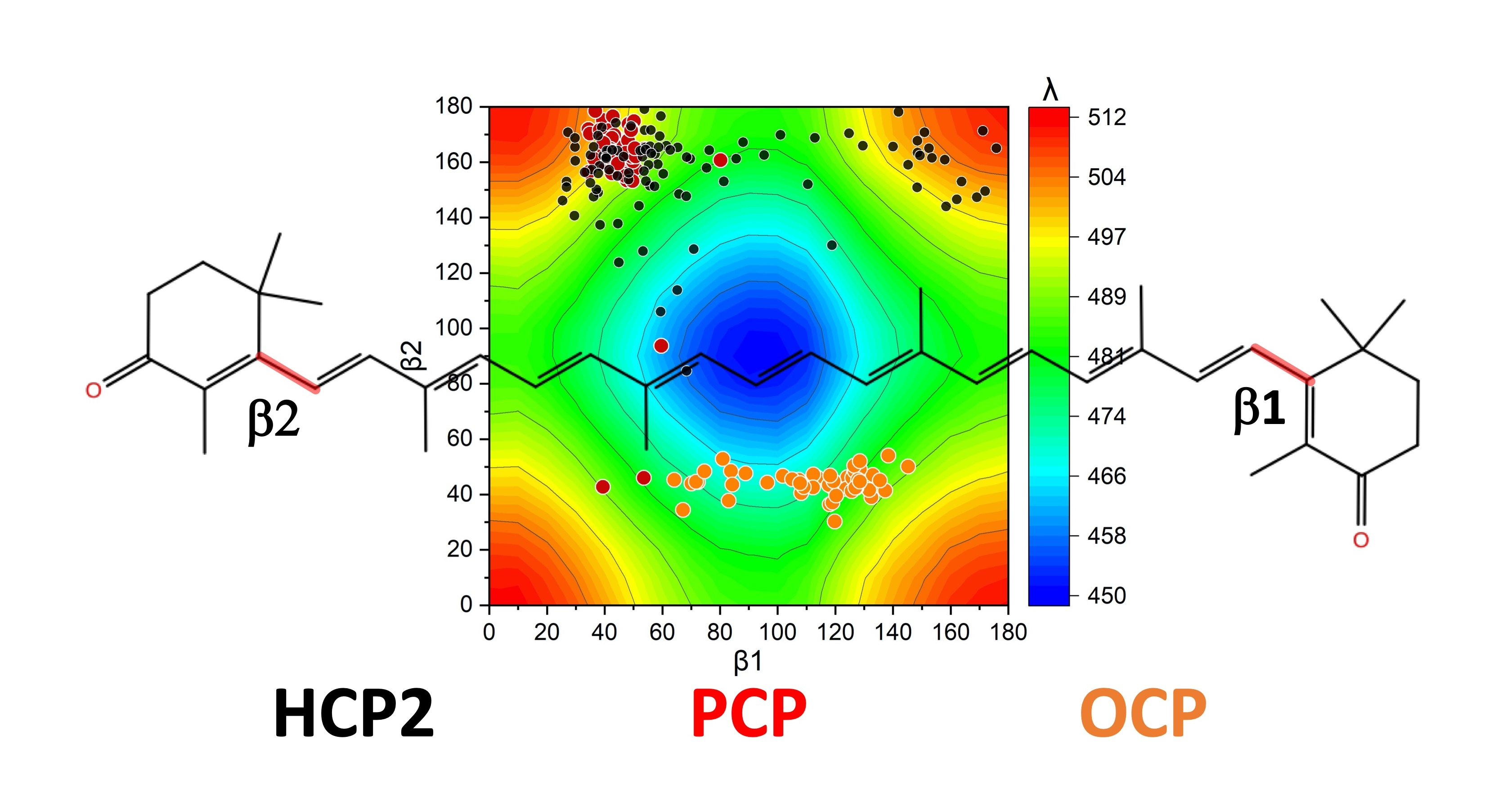
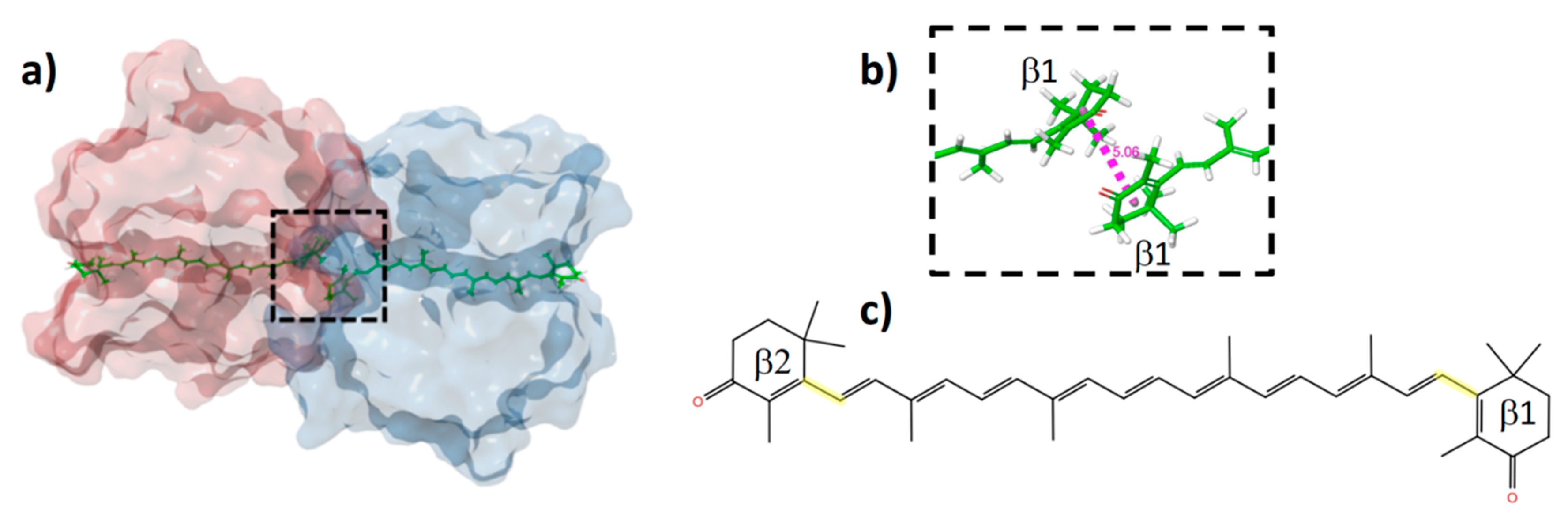
2.2. Distortions, Environmental Influence, and Excitonic Coupling
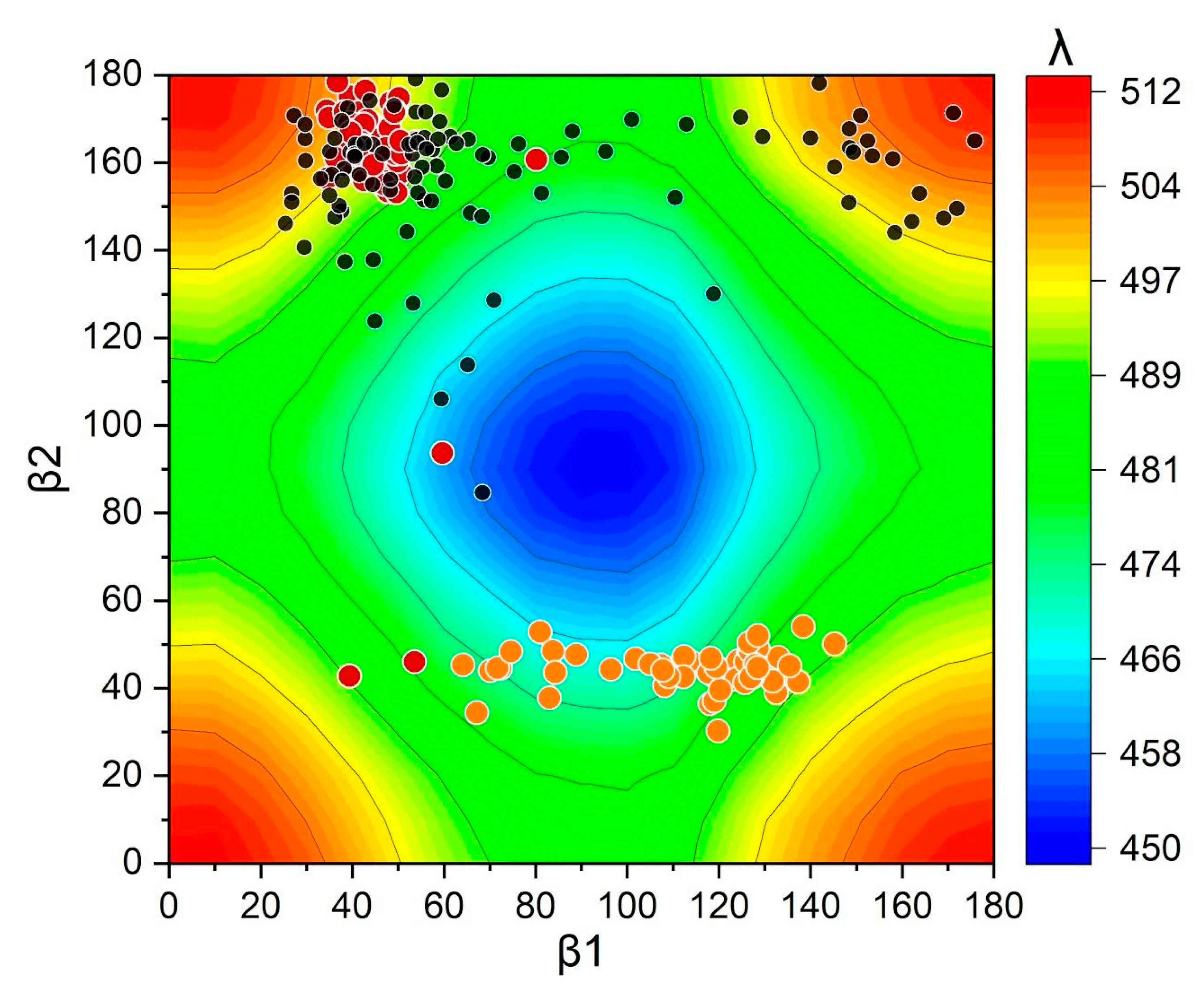
2.3. Molecular Dynamics of HCP2 Monomers
3. Materials and Methods
3.1. Model Preparation
3.2. Structure Optimizations and QM/MM Calculations
3.3. Electronic Coupling Calculations
3.4. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kerfeld, C.A.; Kirilovsky, D. Chapter One—Structural, Mechanistic and Genomic Insights into OCP-Mediated Photoprotection. In Advances in Botanical Research; Chauvat, F., Cassier-Chauvat, C., Eds.; Academic Press: Cambridge, MA, USA, 2013; Volume 65, pp. 1–26. [Google Scholar]
- Steiger, S.; Schäfer, L.; Sandmann, G. High-light-dependent upregulation of carotenoids and their antioxidative properties in the cyanobacterium Synechocystis PCC 6803. J. Photochem. Photobiol. B 1999, 52, 14–18. [Google Scholar] [CrossRef]
- Zhu, Y.; Graham, J.E.; Ludwig, M.; Xiong, W.; Alvey, R.M.; Shen, G.; Bryant, D.A. Roles of xanthophyll carotenoids in protection against photoinhibition and oxidative stress in the cyanobacterium Synechococcus sp. strain PCC 7002. Arch. Biochem. Biophys. 2010, 504, 86–99. [Google Scholar] [CrossRef]
- Kerfeld, C.A.; Melnicki, M.R.; Sutter, M.; Dominguez-Martin, M.A. Structure, function and evolution of the cyanobacterial orange carotenoid protein and its homologs. New Phytol. 2017, 215, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Kerfeld, C.A.; Sawaya, M.R.; Brahmandam, V.; Cascio, D.; Ho, K.K.; Trevithick-Sutton, C.C.; Krogmann, D.W.; Yeates, T.O. The Crystal Structure of a Cyanobacterial Water-Soluble Carotenoid Binding Protein. Structure 2003, 11, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Melnicki, M.R.; Leverenz, R.L.; Sutter, M.; López-Igual, R.; Wilson, A.; Pawlowski, E.G.; Perreau, F.; Kirilovsky, D.; Kerfeld, C.A. Structure, Diversity, and Evolution of a New Family of Soluble Carotenoid-Binding Proteins in Cyanobacteria. Mol. Plant 2016, 9, 1379–1394. [Google Scholar] [CrossRef] [Green Version]
- Muzzopappa, F.; Wilson, A.; Yogarajah, V.; Cot, S.; Perreau, F.; Montigny, C.; Bourcier de Carbon, C.; Kirilovsky, D. The paralogs to the C-terminal domain of the cyanobacterial OCP are carotenoid donors to HCPs. Plant Physiol. 2017. [Google Scholar] [CrossRef]
- Muzzopappa, F.; Wilson, A.; Kirilovsky, D. Interdomain interactions reveal the molecular evolution of the orange carotenoid protein. Nat. Plants 2019, 5, 1076–1086. [Google Scholar] [CrossRef]
- Dominguez-Martin, M.A.; Polívka, T.; Sutter, M.; Ferlez, B.; Lechno-Yossef, S.; Montgomery, B.L.; Kerfeld, C.A. Structural and spectroscopic characterization of HCP2. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 414–424. [Google Scholar] [CrossRef]
- Wei, T.; Balevičius, V.; Polívka, T.; Ruban, A.V.; Duffy, C.D.P. How carotenoid distortions may determine optical properties: Lessons from the Orange Carotenoid Protein. Phys. Chem. Chem. Phys. 2019, 21, 23187–23197. [Google Scholar] [CrossRef] [PubMed]
- Maksimov, E.G.; Shirshin, E.A.; Sluchanko, N.N.; Zlenko, D.V.; Parshina, E.Y.; Tsoraev, G.V.; Klementiev, K.E.; Budylin, G.S.; Schmitt, F.-J.; Friedrich, T.; et al. The Signaling State of Orange Carotenoid Protein. Biophys. J. 2015, 109, 595–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondanza, M.; Cupellini, L.; Lipparini, F.; Mennucci, B. The Multiple Roles of the Protein in the Photoactivation of Orange Carotenoid Protein. Chem 2020, 6, 187–203. [Google Scholar] [CrossRef]
- Khan, T.; Dominguez-Martin, M.A.; Šímová, I.; Fuciman, M.; Kerfeld, C.A.; Polívka, T. Excited-State Properties of Canthaxanthin in Cyanobacterial Carotenoid-Binding Proteins HCP2 and HCP3. J. Phys. Chem. B 2020, 124, 4896–4905. [Google Scholar] [CrossRef] [PubMed]
- Young, A.J.; Phillip, D.M.; Hashimoto, H. Ring-to-chain conformation may be a determining factor in the ability of xanthophylls to bind to the bulk light-harvesting complex of plants. J. Mol. Struct. 2002, 642, 137–145. [Google Scholar] [CrossRef]
- Harms, M.J.; Castañeda, C.A.; Schlessman, J.L.; Sue, G.R.; Isom, D.G.; Cannon, B.R.; García-Moreno, E.B. The pKa Values of Acidic and Basic Residues Buried at the Same Internal Location in a Protein Are Governed by Different Factors. J. Mol. Biol. 2009, 389, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Šlouf, V.; Kuznetsova, V.; Fuciman, M.; de Carbon, C.B.; Wilson, A.; Kirilovsky, D.; Polívka, T. Ultrafast spectroscopy tracks carotenoid configurations in the orange and red carotenoid proteins from cyanobacteria. Photosynth. Res. 2017, 131, 105–117. [Google Scholar] [CrossRef]
- Pigni, N.B.; Clark, K.L.; Beck, W.F.; Gascón, J.A. Spectral Signatures of Canthaxanthin Translocation in the Orange Carotenoid Protein. J. Phys. Chem. B 2020, 124, 11387–11395. [Google Scholar] [CrossRef]
- Gascón, J.A.; Sproviero, E.M.; Batista, V.S. Computational Studies of the Primary Phototransduction Event in Visual Rhodopsin. Acc. Chem. Res. 2006, 39, 184–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curutchet, C.; Mennucci, B. Toward a Molecular Scale Interpretation of Excitation Energy Transfer in Solvated Bichromophoric Systems. J. Am. Chem. Soc. 2005, 127, 16733–16744. [Google Scholar] [CrossRef]
- Russo, V.; Curutchet, C.; Mennucci, B. Towards a Molecular Scale Interpretation of Excitation Energy Transfer in Solvated Bichromophoric Systems. II. The Through-Bond Contribution. J. Phys. Chem. B 2007, 111, 853–863. [Google Scholar] [CrossRef]
- Guberman-Pfeffer, M.J.; Gascón, J.A. Carotenoid-Chlorophyll Interactions in a Photosynthetic Antenna Protein: A Supramolecular QM/MM Approach. Molecules 2018, 23, 2589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guberman-Pfeffer, M.J.; Greco, J.A.; Birge, R.R.; Frank, H.A.; Gascón, J.A. Light Harvesting by Equally Contributing Mechanisms in a Photosynthetic Antenna Protein. J. Phys. Chem. Lett. 2018, 9, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Tilluck, R.W.; Ghosh, S.; Guberman-Pfeffer, M.J.; Roscioli, J.D.; Gurchiek, J.K.; LaFountain, A.M.; Frank, H.A.; Gascón, J.A.; Beck, W.F. Interexciton nonradiative relaxation pathways in the peridinin-chlorophyll protein. Cell Rep. Phys. Sci. 2021, 2, 100380. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Gurchiek, J.K.; Rose, J.B.; Guberman-Pfeffer, M.J.; Tilluck, R.W.; Ghosh, S.; Gascón, J.A.; Beck, W.F. Fluorescence Anisotropy Detection of Barrier Crossing and Ultrafast Conformational Dynamics in the S2 State of β-Carotene. J. Phys. Chem. B 2020, 124, 9029–9046. [Google Scholar] [CrossRef] [PubMed]
- Andreussi, O.; Knecht, S.; Marian, C.M.; Kongsted, J.; Mennucci, B. Carotenoids and Light-Harvesting: From DFT/MRCI to the Tamm–Dancoff Approximation. J. Chem. Theory Comput. 2015, 11, 655–666. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | QM/MM λmax (nm) | β1 Dihedrals | β2 Dihedrals | Experimental λmax (nm) |
|---|---|---|---|---|
| Dimer | 515 | 548 [9] | ||
| Monomer Ligand A | 478 | 74.0° | 179.3° | 530 [9] |
| Monomer Ligand B | 492 | 37.2° | 178.3° | |
| Shift (Dimer–Monomer) | 23 * | 18 | ||
| CAN (in n-hexane) | 465 | 40.7° | 143.3° | 465 [13] |
| CAN (in water) | 470 | 41.4° | 140.3° | N/A |
| Model | Site Energy λmax nm (eV) | EET Coupling (eV) | Eigenvalues (nm) | QM/MM λmax (nm) | Exp. λmax (nm) |
|---|---|---|---|---|---|
| Monomer A | 486 (2.551) | 0.061 | 481 | 484 | 548 |
| Monomer B | 511 (2.427) | 514 | 515 |
| Model | λmax (nm) | Confidence Interval (95%) | SD | Experimental λmax (nm) | |
|---|---|---|---|---|---|
| Monomer Lig. A | 481.9 | 478.3 | 485.5 | 12.7 | 530 |
| Monomer Lig. B | 477.0 | 472.4 | 481.5 | 16.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, K.; Pigni, N.B.; Wijesiri, K.; Gascón, J.A. Spectral Features of Canthaxanthin in HCP2. A QM/MM Approach. Molecules 2021, 26, 2441. https://doi.org/10.3390/molecules26092441
Clark K, Pigni NB, Wijesiri K, Gascón JA. Spectral Features of Canthaxanthin in HCP2. A QM/MM Approach. Molecules. 2021; 26(9):2441. https://doi.org/10.3390/molecules26092441
Chicago/Turabian StyleClark, Kevin, Natalia B. Pigni, Kithmini Wijesiri, and José A. Gascón. 2021. "Spectral Features of Canthaxanthin in HCP2. A QM/MM Approach" Molecules 26, no. 9: 2441. https://doi.org/10.3390/molecules26092441
APA StyleClark, K., Pigni, N. B., Wijesiri, K., & Gascón, J. A. (2021). Spectral Features of Canthaxanthin in HCP2. A QM/MM Approach. Molecules, 26(9), 2441. https://doi.org/10.3390/molecules26092441