
Stereoselective Synthesis of Selenium-Containing Glycoconjugates via the Mitsunobu Reaction



Abstract
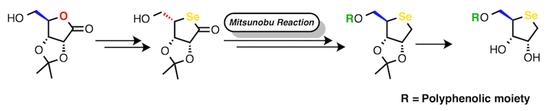
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
- Preparation of 2,3-O-isopropylidene-b-d-ribofuranose (2): To a magnetically stirred suspension of anhydrous triphenylphosphine (0.525 g, ca. 2.0 mmol) in anhydrous acetone (6.0 mL) at r.t., a solution of I2 (0.508 g, 2.0 mmol) in the same solvent (18.0 mL) was added dropwise in the dark and under dry N2 atmosphere. After 15 min, solid d-ribose (0.150 g, 1.0 mmol) was added in one portion to the suspension. TLC monitoring (CHCl3–MeOH, 9:1) showed that the starting sugar was completely consumed within 30 min. The reaction mixture was then filtered, washed with acetone, and dried (Na2SO4). The evaporation of the solvent under reduced pressure gave a crude residue that was used directly in the next step without further purification (0.181 g, 0.95 mmol, 95%). 1H NMR (500 MHz): δ 1.33 (s, 3 H, CH3), 1.48 (s, 3 H, CH3), 3.70 (dd, J5a,5b = 11.6, J5a,4 = 1.5, 1H, H5a), 3.79 (dd, J5b,5a = 11.6, J5b,4 = 1.8, 1 H, H5b), 4.30 (m, 1 H, H4), 4.51 (d, J2,3 = 6.1 Hz, 1 H, H2), 4.84 (dd, J3,2 = 6.1, J3,4 = 1.0 Hz, 1 H, H3), 5.06 (bs, 1 H, H1β). 13C NMR (125 MHz): δ 24.8, 26.4, 65.2, 81.9, 86.5, 87.7, 106.7, 111.9. Anal. Calcd for C8H14O5: C, 50.52; H, 7.42. Found: C, 50.60; H, 7.40. The NMR spectral data were identical to values in the literature [19].
- Preparation of 5-O-tert-butyldiphenylsilyl-2,3-O-isopropylidene-d-ribofuranose (3): TEA (0.28 mL, 2.0 mmol), DMAP (0.024 g, 0.2 mmol), and TBDPSCl (0.26 mL, 1.5 mmol) at 0 °C were added to a magnetically stirred solution of 2 (0.190 g, 1.0 mmol) in anhydrous CH2Cl2 (5.0 mL). The mixture was stirred at room temperature for 4 h. As soon as the starting compound 2 was completely consumed (TLC monitoring), the organic layer was washed with brine and dried (Na2SO4). The evaporation of the solvent under reduced pressure gave a crude residue that was purified by silica gel column chromatography (hexane-ethyl acetate 97:3 (v/v)) to give the oily product 3 as a yellowish syrup (0.386 g, 0.90 mmol, 90%). The product was obtained as a mixture of both anomers, but the βanomer was the main product (α:β, 1:1.9). 1H NMR (500 MHz): δ 1.08 (s, 9Hα, CH3), 1.12 (s, 9Hβ,CH3), 1.34 (s, 3Hβ,CH3), 1.42 (s, 3Hα CH3), 1.50 (s, 3H, 3Hβ,CH3), 1.58 (s, 3Hα, CH3), 3.71–3.63 (m, 2H, H5aα+β), 3.87–3.81 (m, 2H, H5bα+β), 3.96 (d, JOH,1α = 11.4 Hz, 1H, OHα), 4.18 (m, 1H, H4α), 4.31 (m, 1H, H4 β),4.51 (d, JOH,1β = 10.6 Hz, 1H, OHβ), 4.63 (d, J2,3 = 5.9 Hz, 1H, H2β), 4.68 (dd, J2,3 = 6.2 Hz, J2,1 = 4.0 Hz, 1H, H2α), 4.74 (d, J3,2 = 5.9 Hz, 1H, H3β), 4.80 (d, J3,2 = 6.2 Hz, 1H, H3α), 5.37 (d, J1,OH = 10.6 Hz, 1H, H1β), 5.64 (dd, J1,OH = 11.4 Hz, J1,2 = 4.0 Hz, 1H, H1α), 7.53–7.64 (m, 20H, Arα+β); 13C NMR (125 MHz): δ 19.1, 22.6, 25.0, 26.5, 27.0, 65.5, 66.1, 81.6, 87.1, 87.4, 98.0, 103.4, 127.7, 127.9, 128.0, 129.7, 130.2, 130.4, 134.8, 135.6, 135.7. Anal. Calcd for C24H32O5Si: C, 67.26; H, 5.53. Found: C, 67.31; H, 5.63.
- Preparation of 5-O-tert-butyldiphenylsilyl-2,3-O-isopropylidene-d-ribitol (4): To a magnetically stirred solution of 3 (0.429 g, 1.0 mmol) in MeOH (10.0 mL), NaBH4 (0.113 g, 4.0 mmol) at 0 °C was added. The mixture was stirred at room temperature for 2.5 h. As soon as the starting compound 3 was completely consumed (TLC monitoring), the solvent was evaporated under reduced pressure and replaced with AcOEt. The organic layer was washed with brine and dried (Na2SO4). The evaporation of the solvent under reduced pressure gave the pure product 4 as a colorless syrup (0.430 g, 1.0 mmol, >99%). 1H NMR (400 MHz): δ 1.10 (s, 9H, CH3), 1.31 (s, 3H, CH3), 1.33 (s, 3H, CH3), 3.10 (m, 2H, H4 and OH), 3.77–3.93 (m, 4H, H1 and H5), 4.14 (m, 1H, H2), 4.38 (m, 1H, H3), 7.40–7.46 (m, 6H, Ar), 7.67–7.70 (m, 4H, Ar); 13C NMR (125 MHz): δ 19.3, 25.2, 26.9, 27.7, 60.9, 65.3, 69.7, 76.5, 77.6, 108.5, 127.8, 130.0, 132.8, 132.9, 135.6. Anal. Calcd for C24H34O5Si: C, 66.94; H, 7.96. Found: C, 66.91; H, 7.93.
- Preparation of 5-O-tert-butyldiphenylsilyl-2,3-O-isopropylidene-1,4-di-O-methanesulfonyl-d-ribitol (5): DMAP (0.002 g, 0.02 mmol), TEA (1.1 mL, 8.0 mmol), and mesylchloride (0.309 mL, 4.0 mmol) at 0 °C were added to a magnetically stirred solution of 4 (0.430 g, 1.0 mmol) in CH2Cl2 (14.0 mL), and the mixture was stirred at room temperature overnight. The organic layer was washed with brine and dried (Na2SO4). The evaporation of the solvent under reduced pressure gave a crude residue that was purified by silica gel column chromatography (hexane-ethyl acetate 6:4 (v/v)) to give the oily product 5 as a yellowish syrup (0.575 g, 0.98 mmol, 98%). 1H NMR (500 MHz): δ 1.11 (s, 9H, CH3), 1.38 (s, 3H, CH3), 1.43 (s, 3H, CH3), 3.06 (s, 6H, CH3), 4.00 (dd, J5a,5b = 12.2 Hz, J5a,4 = 3.8 Hz, 1H, H5a), 4.07 (dd, J5b,5a = 12.2 Hz, J5b,4 = 2.7 Hz, 1H, H5b), 4.34 (dd, J1a,1b = 10.4 Hz, J1a,2 = 6.4 Hz, 1H, H1a), 4.46–4.54 (m, 3H, H1b, H2 and H3), 4.85 (m, 1H, H4), 7.40–7.49 (m, 6H, Ar), 7.73–7.67 (m, 4H, Ar); 13C NMR (125 MHz): δ 19.3, 25.4, 26.8, 27.4, 37.5, 39.3, 63.1, 68.3, 73.8, 74.9, 79.2, 109.5, 127.9, 130.1, 132.4, 132.5, 135.6. Anal. Calcd for C26H38O9S2Si: C, 53.22; H, 6.53. Found: C, 53.23; H, 6.43.
- Preparation of 5-O-tert-butyldiphenylsilyl-1,4-deoxy-2,3-O-isopropylidene-4-seleno-l-lyxose (6): NaBH4 was added to a magnetically stirred suspension of selenium powder (0.166 g, 2.1 mmol) in dry ethanol (13 mL) under N2 atmosphere, and the mixture was stirred at room temperature until the color of the mixture changed from black to colorless. Then, compound 5 (0.586 g, 1 mmol) in dry THF (8 mL) was added to the mixture, which was heated at 60 °C overnight. Then, the solvent was removed and replaced with AcOEt. Next, the organic layer was washed with brine and dried (Na2SO4). The evaporation of the solvent under reduced pressure gave the pure seleno derivative 6 as a pale-yellow oil (0.470 g, 0.99 mmol, 99%). 1H NMR (500 MHz): δ 1.08 (s, 9H, CH3), 1.30 (s, 3H, CH3), 1.48 (s, 3H, CH3), 2.97 (dd, J1a,1b = 12.2 Hz, J1a,2 = 1.5 Hz, 1H, H1a), 3.03 (dd, J1b,1a = 12.2 Hz, J1b,2 = 4.7 Hz, 1H, H1b), 3.77 (m, 1H, H4), 3.93 (dd, J5a,5b = 10.2 Hz, J5a,4 = 7.9 Hz, 1H, H5a), 4.15 (dd, J5b,5a = 10.2 Hz, J5b,4 = 7.1 Hz, 1H, H5b), 4.77 (m, 1H, H2), 4.94 (dd, J3,2 = 5.3 Hz, J3,4 = 4.5 Hz, 1H, H3), 7.37–7.47 (m, 6H, Ar), 7.72 (ddd, J = 11.0, 7.9, 1.4 Hz, 4H, Ar); 13C NMR (125 MHz): δ 19.3, 24.7, 26.1, 26.8, 29.8, 50.5, 63.0, 83.3, 84.9, 110.3, 127.6, 127.7, 129.6, 129.7, 133.5, 133.6, 135.7, 135.7. Anal. Calcd for C24H32O3SeSi: C, 60.62; H, 6.78. Found: C, 60.59; H, 6.83.
- Under the above conditions, 5-O-tert-butyldiphenylsilyl-1,4-deoxy-2,3-O-isopropylidene-4-seleno-d-ribose (10) was obtained as a pale-yellow oil (96%) starting from the C4 isomer of compound 5: 1H NMR (400 MHz): δ 1.09 (s, 9H, CH3), 1.33 (s, 3H, CH3), 1.54 (s, 3H, CH3), 2.96 (dd, J1a,1b = 11.8 Hz, J1a,2 = 2.3 Hz, 1, H, H1a), 3.20 (dd, J1b,1a = 11.8 Hz, J1b,2 = 5.1 Hz, 1, H, H1b), 3.67 (ddt, J4,5a = 7.3 Hz, J4,5b = 5.3 Hz, J4,3 = 1.8 Hz, 1H, H4), 3.74 (dd, J5a,5b = 10.5 Hz, J5a,4 = 7.0 Hz, 1H, H5a), 3.88 (dd, J5b,5a = 10.5 Hz, J5b,4 = 5.4 Hz, 1H, H5b), 4.80 (dd, J3,2 = 5.3 Hz, J3,4 = 1.8 Hz, 1H, H3), 4.86 (ddd, J2,1b = 5.5 Hz, J2,3 = 5.4 Hz, J2,1a = 2.3 Hz, 1H, H2), 7.38–7.47 (m, 6H, Ar), 7.65–7.72 (m, 4H, Ar); 13C NMR (100 MHz): δ 19.2, 24.7, 26.9, 29.6, 50.1, 64.5, 85.1, 87.3, 110.3, 127.7, 129.8, 133.1, 135.6. Anal. Calcd for C24H32O3SeSi: C, 60.62; H, 6.78. Found: C, 60.63; H, 6.79.
- Preparation of 1,4-deoxy-2,3-O-isopropylidene-4-seleno-l-lyxose (7): A solution of tetrabutylammonium fluoride (1.7 mL of a 1.1 M solution in THF, 2.0 mmol) was added to a magnetically stirred solution of seleno derivative 6 (0.475 g, 1.0 mmol) in anhydrous THF (7.0 mL) at 0 °C under N2, and the mixture was stirred at room temperature for 1 h. The evaporation of the solvent under reduced pressure gave a crude residue that was purified by silica gel column chromatography (hexane-ethyl acetate 6:4 (v/v)) to give the pure product 7 as a crystalline white solid (0.216 g, 0.91 mmol, 91%). 1H NMR (500 MHz): δ 1.33 (s, 3H, CH3), 1.53 (s, 3H, CH3), 2.53 (brs, 1H, OH), 2.99–3.05 (m, 2H, H1), 3.66 (dt, J4,5b = 7.5 Hz, J4,5a = 5.0 Hz, J4,3 = 4.2 Hz, 1H, H4), 3.91 (dd, J5a,5b = 11.5 Hz, J5a,4 = 5.0 Hz, 1H, H5a), 4.00 (dd, J5b,5a = 11.5 Hz, J5b,4 = 7.5 Hz, 1H, H5b), 4.87 (m, 1H, H3), 4.94 (ddd, J2,3 = 6.1 Hz, J2,1b = 4.1 Hz, J2,1a = 2.0 Hz, 1H, H2); 13C NMR (125 MHz): δ 24.5, 26.1, 29.0, 48.2, 62.2, 85.0, 85.1, 111.5. Anal. Calcd for C8H14O3Se: C, 40.52; H, 5.95. Found: C, 40.49; H, 5.96.
- Under the above conditions, 1,4-deoxy-2,3-O-isopropylidene-4-seleno-d-ribose (11) was obtained as an amorphous solid (90%) starting from the C4 isomer of compound 10. 1H NMR (500 MHz): δ 1.33 (s, 3H, CH3), 1.54 (s, 3H, CH3), 3.02 (dd, J1a,1b = 12.0, J1a,2 = 2.1 Hz, 1H, H1a), 3.22 (dd, J1b,1a = 12.0 Hz, J1b,2 = 5.2, 1H, H1b), 3.62–3.75 (m, 3H, H4 and H5), 4.73 (dd, J3,2 = 5.5 Hz, J3,4 = 1.4 Hz, 1H, H3), 4.99 (ddd, J2,3 = 5.5 Hz, J2,1b = 5.2 Hz, J2,1a = 2.1 Hz, 1H, H2); 13C NMR (125 MHz): δ 24.7, 26.8, 29.4, 52.2, 64.1, 85.1, 87.7, 111.6. Anal. Calcd for C8H14O3Se: C, 40.52; H, 5.95. Found: C, 40.55; H, 5.93.
- Preparation of monoacetyl hydroquinone: DMAP (0.01 g, 0.091 mmol) was added to a magnetically stirred solution of hydroquinone (0.100 g, 0.908 mmol) in acetic anhydride (1.09 mmol, 0.103 mL) and dry pyridine (0.90 mL) under N2. The reaction mixture was stirred overnight at room temperature and then diluted in ethyl acetate (50 mL). The resulting solution was washed with HCl (1 M, 50 mL × 4), NH4Cl (50 mL), and brine and dried (Na2SO4). The evaporation of the solvent under reduced pressure gave a crude residue that was purified by silica gel column chromatography (CHCl3) to give the pure monoacetylated hydroquinone as a crystalline white solid (0.125 g, 0.82 mmol, 70%). The NMR spectral data were identical to the values in the literature [25].
- Mitsunobu Reaction: Preparation of glycoconjugates. Typical Procedure: DIAD (0.303 mL, 1.5 mmol) was added to a magnetically stirred solution of TPP (0.393 g, 1.5 mmol) in anhydrous THF (2.3 mL) at 0 °C under N2. After 15 min, a solution containing seleno derivative 7 (0.200 g, 1.0 mmol) and monoacetylated hydroquinone (0.270 g, 1.5 mmol) in anhydrous THF (3.0 mL) was added dropwise. The reaction mixture was stirred at room temperature for three days. The solvent was evaporated under reduced pressure and replaced with AcOEt. The organic layer was washed with brine and dried (Na2SO4). The evaporation of the solvent under reduced pressure gave a crude residue that was purified by silica gel column chromatography (hexane-diethyl ether 7:3 (v/v)) to give the pure product 8 as a pale yellow-syrup (0.160 g, 0.43 mmol, 43%). 1H NMR (500 MHz): δ 1.36 (s, 3H, CH3), 1.57 (s, 3H, CH3), 2.30 (s, 3H, CH3), 3.08 (dd, J1a,1b = 12.0 Hz, J1a,2 = 1.5 Hz, 1H, H1a), 3.34 (dd, J1b,1a = 12.0 Hz, J1b,2 = 5.0 Hz, 1H, H1b), 3.85 (m, 1H, H4), 4.03 (dd, J5a,5b = 9.5 Hz, J5a,4 = 8.0 Hz, 1H, H5a), 4.21 (dd, J5b,5a = 9.5 Hz, J5b,4 = 5.5 Hz, 1H, H5b), 4.90 (dd, J3,2 = 5.5 Hz, J3,4 = 1.5 Hz, 1H, H3), 5.05 (dt, J2,3 = 5.5 Hz, J2,1b = 5.0 Hz, J2,1a = 2.0 Hz, 1H, H2), 6.90 (d, J = 9.0 Hz, 2H, Ar), 7.00 (d, J = 9.0 Hz, 2H, Ar); 13C NMR (125 MHz): δ 21.0, 24.7, 26.7, 30.2, 47.0, 70.9, 85.2, 87.6, 110.6, 115.3, 122.4, 144.6, 156.0, 169.8. Anal. Calcd for C16H20O5Se: C, 51.76; H, 5.43. Found: C, 51.66; H, 5.48.
- Under the above conditions, 5-(4-hydroxyphenoxy)-1,4-deoxy-2,3-O-isopropylidene-4-seleno-d-ribose (12) was obtained as oil (13%) starting from the compound 7 and hydroquinone: 1H NMR (500 MHz): δ 1.36 (s, 3H, CH3), 1.56 (s, 3H, CH3), 3.06 (dd, J1a,1b = 11.9 Hz, J1a,2 = 1.9 Hz, 1H, H1a), 3.33 (dd, J1b,1a = 11.9 Hz, J1b,2 = 5.1 Hz, 1H, H1b), 3.83 (m, 1H, H4), 3.99 (dd, J5a,5b = 9.9 Hz, J5a,4 = 7.8 Hz, 1H, H5a), 4.16 (dd, J5b,5a = 9.9 Hz, J5b,4 = 5.2 Hz, 1H, H5b), 4.91 (dd, J3,2 = 5.6 Hz, J3,4 = 1.7 Hz, 1H, H3), 5.05 (dt, J2,3 = 5.6 Hz, J2,1b = 5.3 Hz, J2,1a = 1.9 Hz, 1H, H2), 6.77 (d, J = 9.2 Hz, 2H, Ar), 6.80 (d, J = 9.3 Hz, 2H, Ar); 13C NMR (125 MHz): δ 24.7, 26.7, 30.2, 47.2, 71.3, 85.2, 87.7, 110.6, 115.9, 116.1, 150.0, 152.5. Anal. Calcd for C14H18O4Se: C, 51.07; H, 5.51. Found: C, 51.10; H, 5.50.
- Under the above conditions, (E)-5-((3-(3,4-dihydroxyphenyl)acryloyl)oxy)-1,4-deoxy-2,3-O-isopropylidene-4-seleno-d-ribose (13) was obtained as oil (70%) starting from the compound 7 and caffeic acid: 1H NMR (500 MHz): δ 1.34 (s, 3H, CH3), 1.54 (s, 3H, CH3), 3.06 (dd, J1a,1b = 12.1 Hz, J1a,2 = 1.7 Hz, 1H, H1a), 3.26 (dd, J1b,1a = 12.1 Hz, J1b,2 = 4.9 Hz, 1H, H1b), 3.76 (m, 1H, H4), 4.26 (dd, J5a,5b = 11.6 Hz, J5a,4 = 8.9 Hz, 1H, H5a), 4.37 (dd, J5b,5a = 11.6 Hz, J5b,4 = 5.8 Hz, 1H, H5b), 4.77 (dd, J3,2 = 5.5 Hz, J3,4 = 1.6 Hz, 1H, H3), 5.03 (ddd, J2,3 = 5.7 Hz, J2,1b = 5.0 Hz, J2,1a = 2.0 Hz, 1H, H2), 6.00 (brs, 2H, OH), 6.24 (d, J = 15.9 Hz, 1H, H2′), 6.87 (d, J = 8.2 Hz, 1H, H8′), 7.00 (dd, J = 8.2 Hz, J = 1.5 Hz, 1H, H9′), 7.07 (d, J = 1.5 Hz, 1H, H5′), 7.58 (d, J = 15.9 Hz, 1H, H3′); 13C NMR (125 MHz): δ 24.7, 26.7, 29.7, 46.6, 65.7, 85.0, 87.3, 110.9, 114.5, 115.1, 115.6, 122.7, 127.6, 143.8, 145.5, 146.3, 167.0. Anal. Calcd for C17H20O6Se: C, 51.14; H, 5.05. Found: C, 51.19; H, 5.03.
- Under the above conditions, (E)-5-((3-(4-hydroxy-3-methoxyphenyl)acryloyl)oxy)-1,4-deoxy-2,3-O-isopropylidene-4-seleno-d-ribose (14) was obtained as oil (68%) starting from the compound 7 and ferulic acid: 1H NMR (500 MHz, acetone-d6): δ 1.30 (s, 3H, CH3), 1.46 (s, 3H, CH3), 2.99 (dd, J1a,1b = 11.9 Hz, J1a,2 = 1.3 Hz, 1H, H1a), 3.37 (dd, J1b,1a = 11.9 Hz, J1b,2 = 4.9 Hz, 1H, H1b), 3.76 (ddd, J4,5a = 8.7 Hz, J4,5b = 7.4 Hz, J4,3 = 1.3 Hz, 1H, H4), 3.95 (s, 3H, CH3), 4.26 (dd, J5a,5b = 11.4 Hz, J5a,4 = 8.8 Hz, 1H, H5a), 4.36 (dd, J5b,5a = 11.4 Hz, J5b,4 = 6.8 Hz, 1H, H5b), 4.86 (dd, J3,2 = 5.8 Hz, J3,4 = 1.5 Hz, 1H, H3), 5.11 (ddd, J2,3 = 6.1, J2,1b = 5.9 Hz, J2,1a = 1.9 Hz, 1H, H2), 6.46 (d, J = 16.0 Hz, 1H, H2′), 6.90 (dd, J = 8.0 Hz, 1H, H8′), 7.18 (dd, J = 8.5 Hz, J = 1.9 Hz, 1H, H9′), 7.39 (d, J = 1.5 Hz, 1H, H5′), 7.66 (d, J = 16.0 Hz, 1H, H3′); 13C NMR (125 MHz): δ 24.7, 26.7, 29.3, 46.7, 56.0, 65.6, 85.0, 87.3, 109.4, 110.8, 114.8, 114.8, 123.3, 126.8, 145.7, 146.8, 148.2, 166.9. Anal. Calcd for C18H22O6Se: C, 52.31; H, 5.37. Found: C, 52.25; H, 5.43.
- Under the above conditions, 5-(4-((1E,6E)-7-(4-hydroxy-3-methoxyphenyl)-3,5-dioxohepta-1,6-dien-1-yl)-2-methoxyphenoxy)-1,4-deoxy-2,3-O-isopropylidene-4-seleno-d-ribose (15) was obtained as oil (30%) starting from the compound 7 and curcumin: 1H NMR (400 MHz): δ 1.36 (s, 3H, CH3), 1.57 (s, 3H, CH3), 3.04 (dd, J1a,1b = 12.0 Hz, J1a,2 = 1.9 Hz, 1H, H1a), 3.55 (dd, J1b,1a = 12.0 Hz, J1b,2 = 5.0 Hz, 1H, H1b), 3.93–3.86 (m, 4H, H4 and CH3), 3.96 (s, 3H, CH3), 4.16 (dd, J5a,5b = 9.7 Hz, J5a,4 = 8.0 Hz, 1H, H5a), 4.33 (dd, J5b,5a = 9.7 Hz, J5b,4 = 5.2 Hz, 1H, H5b), 4.95 (dd, J3,2 = 5.6 Hz, J3,4 = 1.8 Hz, 1H, H3), 5.11 (ddd, J2,3 = 5.6 Hz, J2,1b = 5.0 Hz, J2,1a = 1.9 Hz, 1H, H2), 5.82 (m, 2H, H10′), 5.98 (bs, 1H, OH), 6.52–6.46 (m, 2H, H8′ and H12′), 6.84 (d, J = 8.3 Hz, 1H, H6′), 6.94 (d, J = 8.3 Hz, 1H, H18′), 7.05 (d, J = 1.7 Hz, 1H, H15′), 7.07 (d, J = 1.9 Hz, 1H, H3′), 7.14–7.09 (m, 2H, H5′ and H19′), 7.65–7.561 (m, 2H, H7′ and H13′); 13C NMR (125 MHz): δ 24.6, 26.7, 31.1, 47.1, 56.0, 72.1, 85.6, 88.2, 101.2, 109.6, 110.4, 113.1, 114.8, 121.8, 122.3, 122.9, 129.0, 140.2, 140.6, 146.8, 147.9, 149.7, 182.9, 183.6. Anal. Calcd for C29H32O8Se: C, 59.29; H, 5.49. Found: C, 59.19; H, 5.55.
- Removal of the O-isopropylidene group. Typical Procedure: A solution (3.3 mL) of CH3COOH/H2O (8:2, v/v) was added to compound 12 (0.329 g, 1.0 mmol). The mixture was stirred at 80 °C for 2 h. Then, the solvent was evaporated under reduced pressure; next, the organic layer was washed with diethylether. The crude residue was purified to afford compound 12a as an amorphus solid (0.205 g, 0.8 mmol, 71%). 1H NMR (400 MHz, acetone-d6): δ 2.82 (dd, J1a,1b = 10.0 Hz, J1a,2 = 5.1 Hz, 1H, H1a), 3.02 (dd, J1b,1a = 10.0 Hz, J1b,2 = 5.0 Hz, 1H, H1b), 3.77 (m, 1H, H4), 3.99 (dd, J5a,5b = 9.7 Hz, J5a,4 = 8.3 Hz, 1H, H5a), 4.10 (m, 1H, H3), 4.35 (dd, J5b,5a = 9.7 Hz, J5b,4 = 5.9 Hz, 1H, H5b), 4.40 (m, 1H, H2), 6.75 (d, J = 9.2 Hz, 2H, Ar), 6.79 (d, J = 9.2 Hz, 2H, Ar); 13C NMR (100 MHz, acetone-d6): δ 23.9, 42.6, 72.4, 76.0, 78.4, 115.8, 151.6, 152.0. Anal. Calcd for C11H14O4Se: C, 45.69; H, 4.88. Found: C, 45.73; H, 4.83.
- Under the above conditions, (E)-5-((3-(3,4-dihydroxyphenyl)acryloyl)oxy)-1,4-deoxy-4-seleno-d-ribose (13a) was obtained as an amorphous solid (68%) starting from the compound 13: 1H NMR (500 MHz, acetone-d6): δ 2.84 (dd, J1a,1b = 9.9 Hz, J1a,2 = 5.3 Hz, 1H, H1a), 3.06 (dd, J1b,1a = 9.9 Hz, J1b,2 = 5.1 Hz, 1H, H1b), 3.74 (m, 1H, H4), 4.10 (dd, J3,4 = 5.9 Hz, J3,2 = 3.2 Hz, 1H, H3), 4.22 (dd, J5a,5b = 11.3 Hz, J5a,4 = 7.6 Hz, 1H, H5a), 4.43 (ddt, J2,1a = 5.3 Hz, J2,1b = 5.1 Hz, J2,3 = 3.1 Hz, 1H, H2), 4.57 (dd, J5b,5a = 11.3 Hz, J5b,4 = 6.6 Hz, 1H, H5b), 6.32 (d, J = 15.9 Hz, 1H, H2′), 6.90 (d, J = 8.2 Hz, 1H, H5′), 7.09 (dd, J = 8.2 Hz, J = 2.0 Hz, 1H, H9′), 7.19 (d, J = 2.0 Hz, 1H, H8′), 7.58 (d, J = 15.6 Hz, 1H, H3′); 13C NMR (125 MHz, acetone-d6): δ 24.1, 41.9, 66.5, 76.0, 78.4, 114.4, 115.5, 121.7, 126.7, 145.2, 145.4, 148.0, 166.2. Anal. Calcd for C14H16O6Se: C, 46.81; H, 4.49. Found: C, 46.81; H, 4.45.
- Under the above conditions, (E)-5-((3-(4-hydroxy-3-methoxyphenyl)acryloyl)oxy)-1,4-deoxy-4-seleno-d-ribose (14a), an amorphous solid, was obtained (80%) starting from the compound 14: 1H NMR (500 MHz, acetone-d6): δ 2.85 (dd, J1a,1b = 10.0 Hz, J1a,2 = 5.3 Hz, 1H, H1a), 3.06 (dd, J1b,1a = 10.0 Hz, J1b,2 = 5.0 Hz, 1H, H1b), 3.74 (m, 1H, H4), 3.95 (s, 3H, CH3), 4.09 (m, 1H, H3), 4.22 (dd, J5a,5b = 11.3 Hz, J5a,4 = 7.8 Hz, 1H, H5a), 4.43 (m, 1H, H2), 4.58 (dd, J5b,5a = 11.3 Hz, J5b,4 = 6.5 Hz, 1H, H5b), 6.43 (d, J = 15.9 Hz, 1H, H2′), 6.90 (d, J = 8.1 Hz, 1H, H8′), 7.18 (dd, J = 8.2 Hz, J = 1.7 Hz, 1H, H9′), 7.39 (bs, 1H, H5′), 7.64 (d, J = 15.9 Hz, 1H, H3′); 13C NMR (125 MHz, acetone-d6): δ 24.1, 41.9, 55.5, 66.6, 76.0, 78.4, 110.4, 114.6, 115.2, 123.3, 126.5, 145.2, 145.3, 147.9, 149.3, 166.3. Anal. Calcd for C15H18O6Se: C, 48.27; H, 4.86. Found: C, 48.36; H, 4.82.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, C.; Wood, P.L.; Romani, A.; Valacchi, G.; Squerzanti, M.; Sanz, J.M.; Ortolani, B.; Zuliani, G. Oxidative challenge in Alzheimer’s disease: State of knowledge and future needs. J. Investig. Med. 2016, 64, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D.J. Antioxidant treatment in Alzheimer’s disease. J. Mol. Neurosci. 2003, 21, 1–11. [Google Scholar] [CrossRef]
- Trippier, P.C.; Labby, K.J.; Hawker, D.D.; Mataka, J.J.; Silverman, R.B. Target- and mechanism-based therapeutics for neurodegenerative diseases: Strength in numbers. J. Med. Chem. 2013, 56, 3121–3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, C.W.; Rocha, J.B.T. Toxicology and pharmacology of selenium: Emphasis on synthetic organoselenium compounds. Arch. Toxicol. 2011, 85, 1313–1359. [Google Scholar] [CrossRef]
- Moussaoui, S.; Obinu, M.C.; Daniel, N.; Reibaud, M.; Blanchard, V.; Imperato, A. The antioxidant ebselen prevents neurotoxicity and clinical symptoms in a primate model of parkinson’s disease. Exp. Neurol. 2000, 166, 235–245. [Google Scholar] [CrossRef]
- Aisen, P.S.; Gauthier, S.; Ferris, S.H.; Saumier, D.; Haine, D.; Garceau, D.; Duong, A.; Suhy, J.; Oh, J.; Lau, W.C.; et al. Tramiprosate in mild-to-moderate Alzheimer’s disease—A randomized, double-blind, placebo-controlled, multi-centre study (the Alphase Study). Arch. Med. Sci. 2011, 7, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.C.; Schneider, L.S.; Amato, D.A. Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients With Mild Alzheimer Disease. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro Espíndola, K.M.; Guimarães Ferreira, R.; Mosquera Narvaez, L.E.; Rocha Silva Rosario, A.C.; Machado da Silva, A.H.; Bispo Silva, A.G.; Oliveira Vieira, A.P.; Chagas Monteiro, M. Chemical and pharmacological aspects of caffeic acid and its activity in hepatocarcinoma. Front. Oncol. 2019, 9, 541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Bougie, P.; Halliez, M.; Maïga, S.; Godon, C.; Kervoëlen, C.; Pellat-Deceunynck, C.; Amiot, M. Curcumin induces cell death of the main molecular myeloma subtypes, particularly the poor prognosis subgroups. Cancer Biol. Ther. 2015, 16, 60–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Yao, J.; Li, Y.; Hu, X.; Shao, H.; Tian, X. Anti-inflammatory and antioxidant effects of curcumin on acute lung injury in a rodent model of intestinal ischemia reperfusion by inhibiting the pathway of NF-Kb. Int. J. Clin. Exp. Pathol. 2015, 8, 3451–3459. [Google Scholar]
- Nobili, A.; Latagliata, E.C.; Viscomi, M.T.; Cavallucci, V.; Cutuli, D.; Giacovazzo, G.; Krashia, P.; Rizzo, F.R.; Marino, R.; Federici, M.; et al. Dopamine neuronal loss contributes to memory and reward dysfunction in a model of Alzheimer’s disease. Nat. Commun. 2017, 8, 14727. [Google Scholar] [CrossRef]
- Jain, V.K.; Priyadarsini, K.I. Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Royal Society of Chemistry: Croydon, UK, 2018. [Google Scholar] [CrossRef]
- Inagaki, Y.; Minakawa, N.; Matsuda, A. Synthesis of 4′selenoribo nucleosides. Nucleic Acids Symp. Ser. 2007, 51, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Jayakanthan, K.; Johnston, B.D.; Pinto, B.M. Stereoselective synthesis of 4′-selenonucleosides using the Pummerer glycosylation reaction. Carbohydr. Res. 2008, 343, 1790–1800. [Google Scholar] [CrossRef]
- Jeong, L.S.; Tosh, D.K.; Kim, H.O.; Wang, T.; Hou, X.; Yun, H.S.; Kwon, Y.; Lee, S.K.; Choi, J.; Zhao, L.X. First Synthesis of 4‘-selenonucleosides showing unusual southern conformation. Org. Lett. 2008, 10, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Kim, J.-H.; Lee, H.W.; Alexander, V.; Ahn, H.-C.; Choi, W.J.; Choi, J.; Jeong, L.S. New RNA purine building blocks, 4′-selenopurine nucleosides: First synthesis and unusual mixture of sugar puckerings. Chem. Eur. J. 2013, 19, 5528–5532. [Google Scholar] [CrossRef] [PubMed]
- Pedatella, S.; Guaragna, A.; D’Alonzo, D.; De Nisco, M.; Palumbo, G. Triphenylphosphine polymer-bound/iodine complex: A suitable reagent for the preparation of o-isopropylidene sugar derivatives. Synthesis 2006, 2, 305–308. [Google Scholar] [CrossRef]
- Caputo, R.; Kunz, H.; Mastroianni, D.; Palumbo, G.; Pedatella, S.; Solla, F. Mild Synthesis of Protected α-D-Glycosyl Iodides. Eur. J. Org. Chem. 1999, 3147–3150. [Google Scholar] [CrossRef]
- Sapkota, K.; Roh, E.; Lee, E.; Ha, E.-M.; Yang, J.-H.; Lee, E.-S.; Kwon, Y.; Kim, Y.; Na, Y. Synthesis and anti-melanogenic activity of hydroxyphenyl benzyl ether analogues. Bioorg. Med. Chem. 2011, 19, 2168–2175. [Google Scholar] [CrossRef]
- Taniguchi, T.; Nakano, K.; Baba, R.; Monde, K. Analysis of configuration and conformation of furanose ring in carbohydrate and nucleoside by vibrational circular dichroism. Org. Lett. 2017, 19, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Batra, H.; Moriarty, R.M.; Penmasta, R.; Sharma, V.; Stanciuc, G.; Staszewski, J.P.; Tuladhar, S.M.; Walsh, D.A. A concise, efficient and production-scale synthesis of a protected l-lyxonolactone derivative: An important aldonolactone core. Org. Process Res. Dev. 2006, 10, 484–486. [Google Scholar] [CrossRef]
- Taniike, H.; Inagaki, Y.; Matsuda, A.; Minakawa, N. Practical synthesis of 4’-selenopyrimidine nucleosides using hypervalent iodine. Tetrahedron 2011, 67, 7977–7982. [Google Scholar] [CrossRef]
- Cepanec, I.; Litvic, M. Simple and efficient synthesis of arbutin. Arkivoc 2008, 2, 19–24. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Compound | 3J2,1a | 3J2,1b | 3J3,2 | 3J3,4 |
| 6 | 1.5 | 4.7 | 5.3 | 4.5 |
| 7 | 2.0 | 4.1 | 6.1 | 4.2 |
| 10 | 2.3 | 5.1 | 5.3 | 1.8 |
| 11 | 2.1 | 5.2 | 5.5 | 1.4 |
| Polyphenol | Glycoconjugate | Yield 2 |
 |  | 13 (14) 3 |
 |  | 43 (44) 3 |
 |  | 70 (72) 3 |
 |  | 68 (67) 3 |
 |  | 30 (28) 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serpico, L.; De Nisco, M.; Cermola, F.; Manfra, M.; Pedatella, S. Stereoselective Synthesis of Selenium-Containing Glycoconjugates via the Mitsunobu Reaction. Molecules 2021, 26, 2541. https://doi.org/10.3390/molecules26092541
Serpico L, De Nisco M, Cermola F, Manfra M, Pedatella S. Stereoselective Synthesis of Selenium-Containing Glycoconjugates via the Mitsunobu Reaction. Molecules. 2021; 26(9):2541. https://doi.org/10.3390/molecules26092541
Chicago/Turabian StyleSerpico, Luigia, Mauro De Nisco, Flavio Cermola, Michele Manfra, and Silvana Pedatella. 2021. "Stereoselective Synthesis of Selenium-Containing Glycoconjugates via the Mitsunobu Reaction" Molecules 26, no. 9: 2541. https://doi.org/10.3390/molecules26092541
APA StyleSerpico, L., De Nisco, M., Cermola, F., Manfra, M., & Pedatella, S. (2021). Stereoselective Synthesis of Selenium-Containing Glycoconjugates via the Mitsunobu Reaction. Molecules, 26(9), 2541. https://doi.org/10.3390/molecules26092541