
D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect


Abstract
:
1. Introduction
2. Results
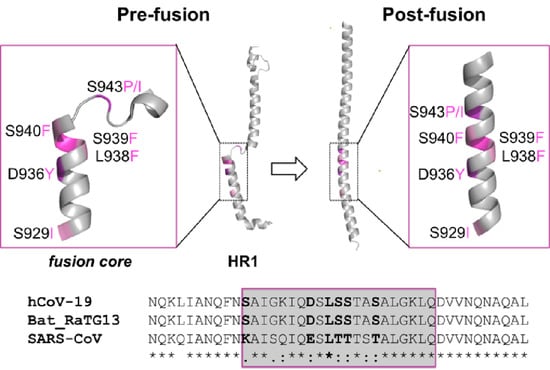
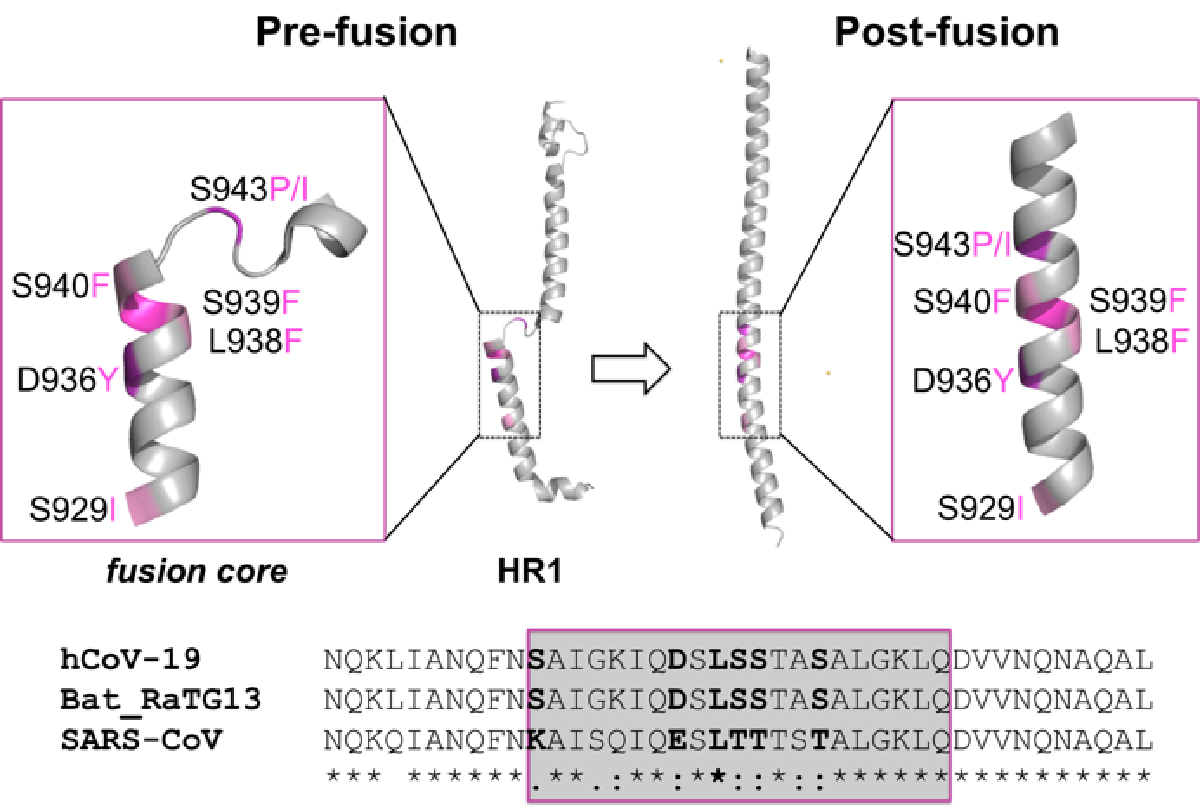
2.1. Identification of the HR1 “Fusion Core” Mutations
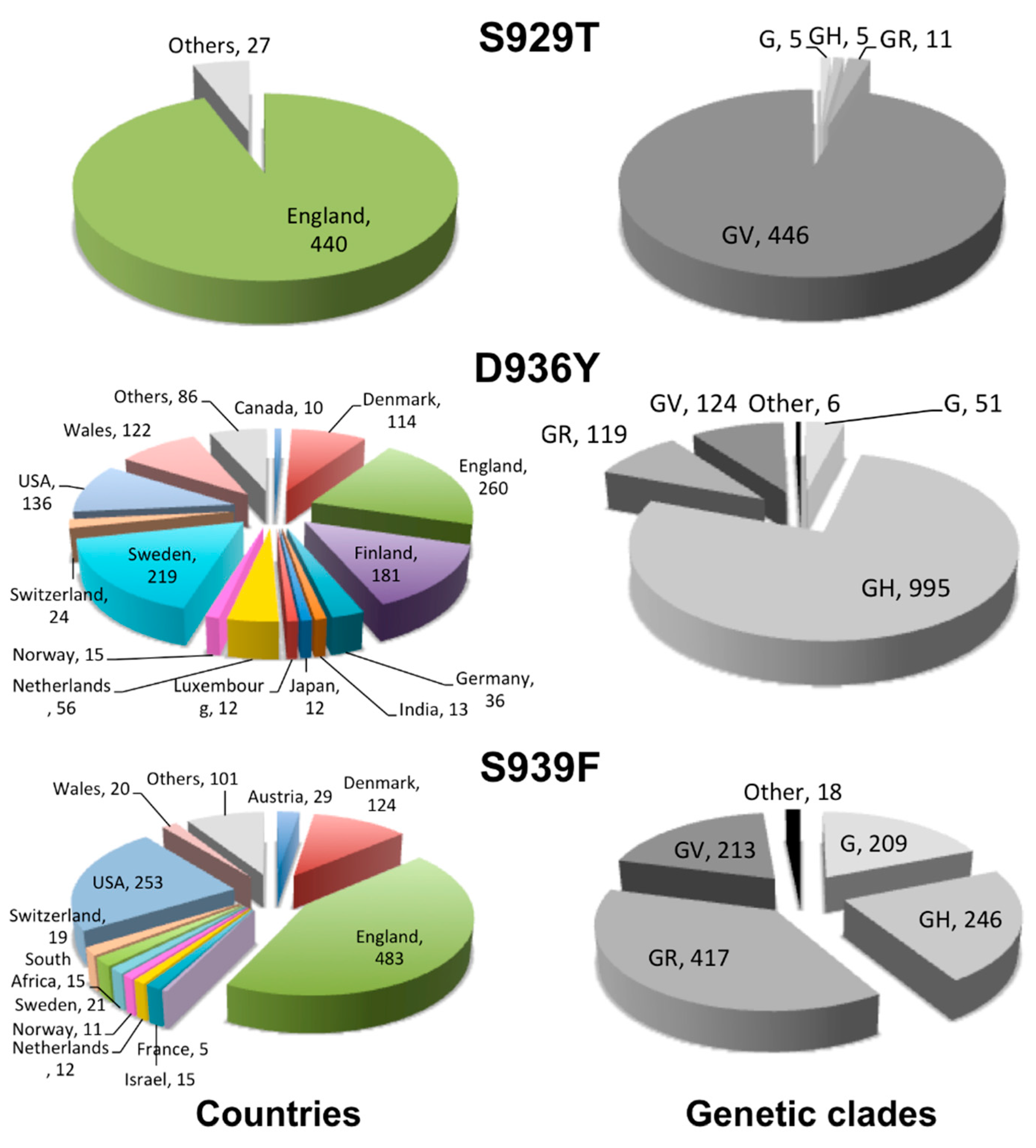
2.2. Geographical Distribution of the HR1 “Fusion Core” of Most Frequent Mutations
2.3. Clade Association of the HR1 “Fusion Core” of Most Frequent Mutations
2.4. Sequence Conservation among Similar Viruses
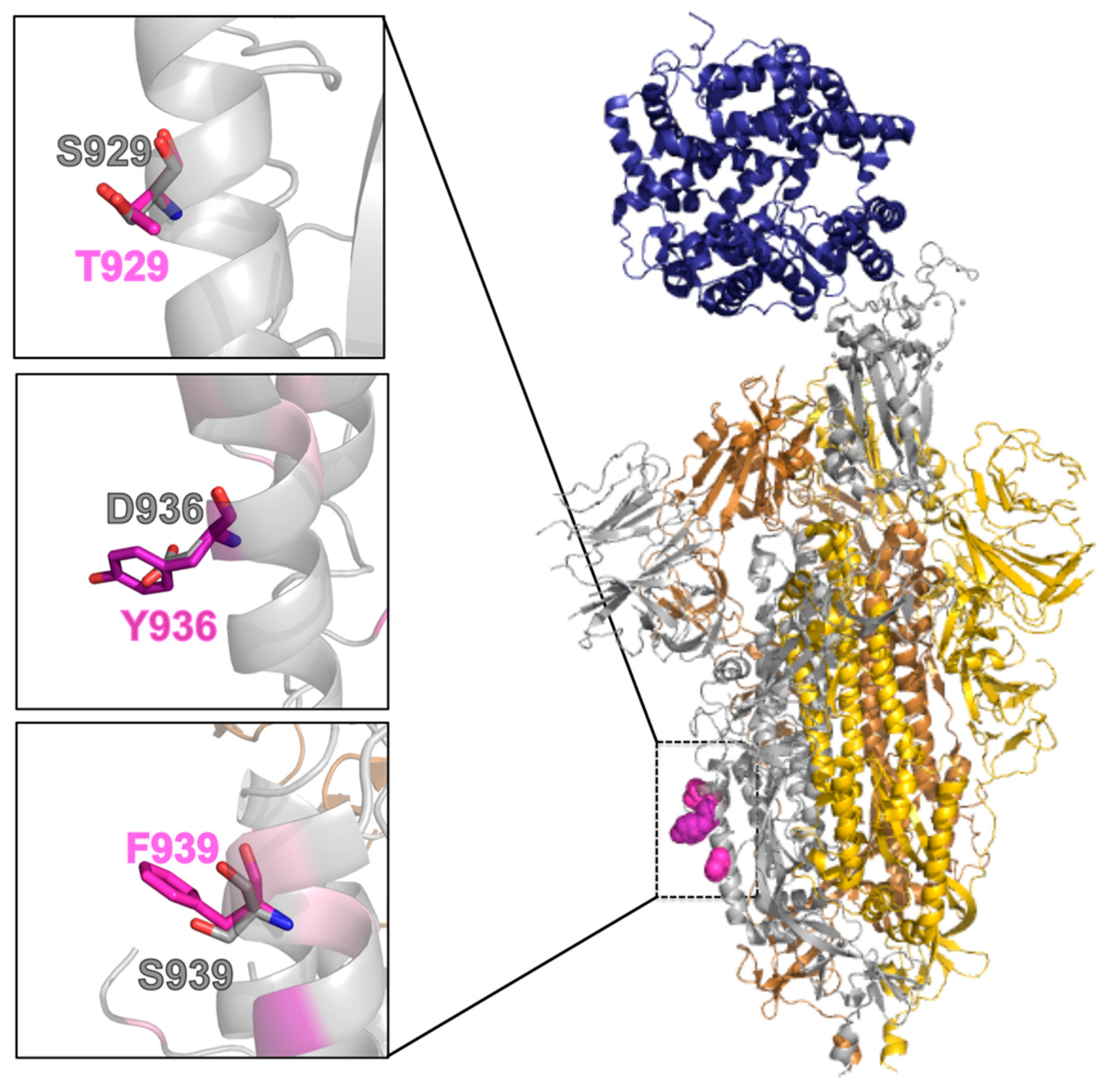
2.5. Effect of the Mutations on the Protein Pre-Fusion Conformation
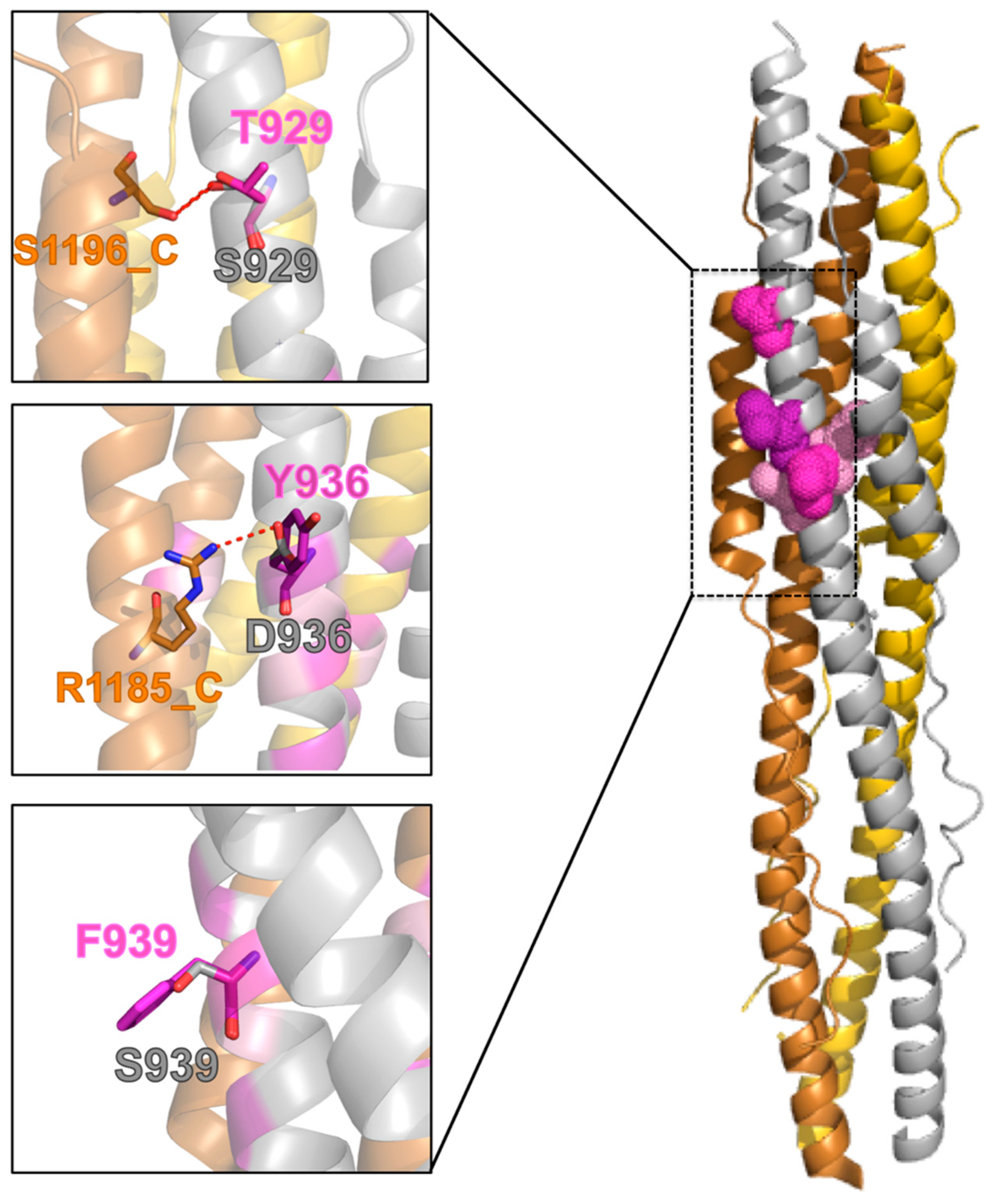
2.6. Effect of the Mutations on the Protein Post-Fusion Conformation
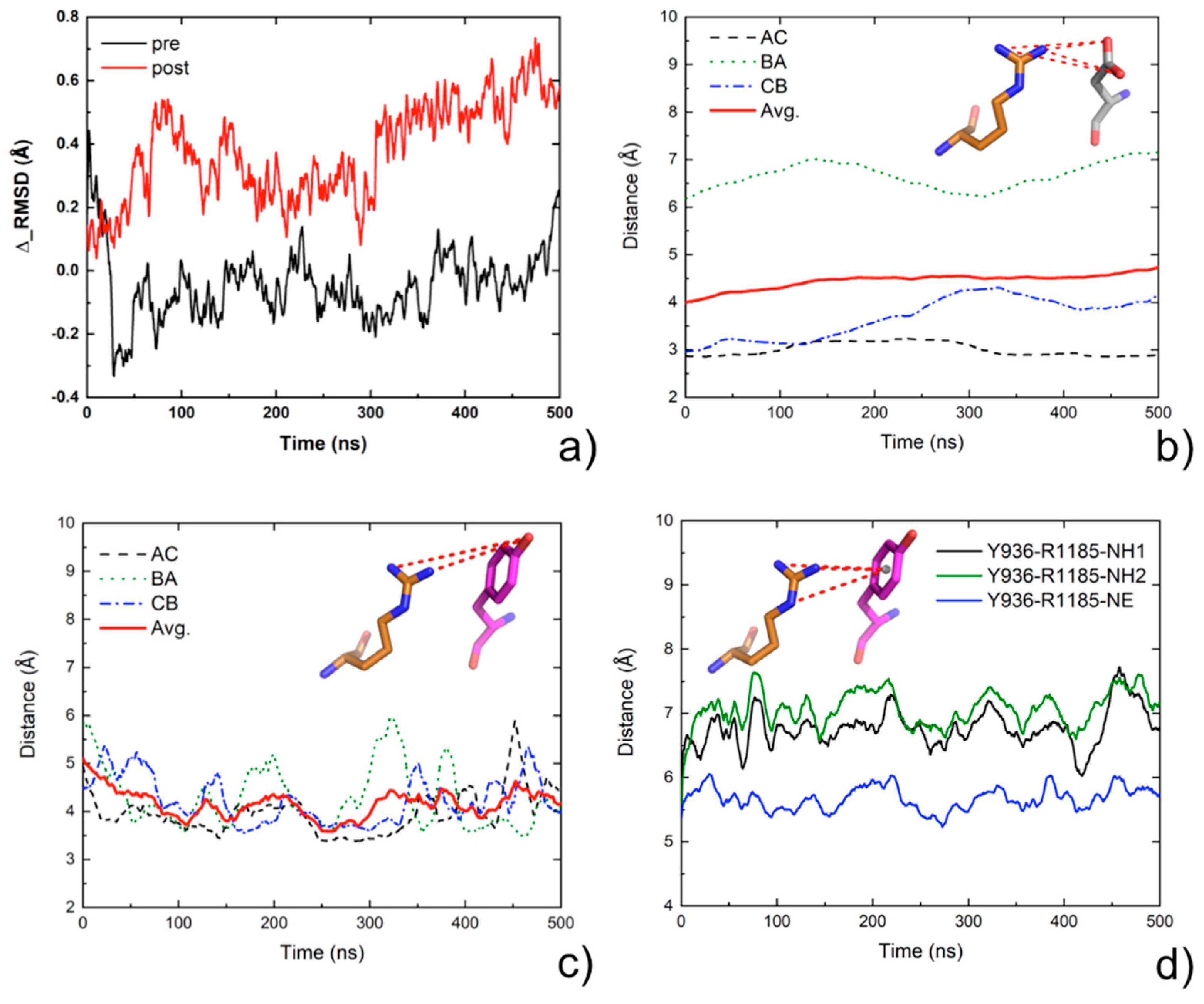
2.7. Molecular Dynamics Analysis
3. Discussion
4. Methods
4.1. Identification of Mutations
4.2. Mutants Modelling and Analysis
4.3. Molecular Dynamics Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Receptor recognition mechanisms of coronaviruses: A decade of structural studies. J. Virol. 2015, 89, 1954–1964. [Google Scholar] [CrossRef] [Green Version]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [Green Version]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Salvatori, G.; Luberto, L.; Maffei, M.; Aurisicchio, L.; Roscilli, G.; Palombo, F.; Marra, E. SARS-CoV-2 SPIKE PROTEIN: An optimal immunological target for vaccines. J. Transl. Med. 2020, 18, 222. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar] [CrossRef]
- McKee, D.L.; Sternberg, A.; Stange, U.; Laufer, S.; Naujokat, C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Krammer, F. SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- Heald-Sargent, T.; Gallagher, T. Ready, set, fuse! The coronavirus spike protein and acquisition of fusion competence. Viruses 2012, 4, 557–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, W.; Farzan, M.; Harrison, S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science 2005, 309, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—from vision to reality. Euro Surveill 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Zhu, Y.; Yu, D.; Yan, H.; Chong, H.; He, Y. Design of Potent Membrane Fusion Inhibitors against SARS-CoV-2, an Emerging Coronavirus with High Fusogenic Activity. J. Virol. 2020, 94, e00620–e00635. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.B.; Li, M.S.; Theodorakis, P.E. Generalization of the elastic network model for the study of large conformational changes in biomolecules. Phys. Chem. Chem. Phys. 2018, 20, 17020–17028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of Structural and Energetic Differences between Conformations of the SARS-CoV-2 Spike Protein. Materials 2020, 13, 5362. [Google Scholar] [CrossRef] [PubMed]
- Alm, E.; Broberg, E.K.; Connor, T.; Hodcroft, E.B.; Komissarov, A.B.; Maurer-Stroh, S.; Melidou, A.; Neher, R.A.; O’Toole, Á.; Pereyaslov, D.; et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Euro Surveill. Bull. Eur. Mal. Transm. Eur. Commun. Dis. Bull. 2020, 25, 2001410. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.E.; Becktel, W.J.; Dahlquist, F.W. pH-induced denaturation of proteins: A single salt bridge contributes 3-5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry 1990, 29, 2403–2408. [Google Scholar] [CrossRef]
- Debiec, K.T.; Gronenborn, A.M.; Chong, L.T. Evaluating the strength of salt bridges: A comparison of current biomolecular force fields. J. Phys. Chem B 2014, 118, 6561–6569. [Google Scholar] [CrossRef] [PubMed]
- Steiner, T.; Koellner, G. Hydrogen bonds with pi-acceptors in proteins: Frequencies and role in stabilizing local 3D structures. J. Mol. Biol 2001, 305, 535–557. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Feyfant, E.; Sali, A.; Fiser, A. Modeling mutations in protein structures. Protein. Sci. 2007, 16, 2030–2041. [Google Scholar] [CrossRef] [Green Version]
- DeLano Scientific, L. 2002. Available online: http://www.pymol.org (accessed on 28 April 2021).
- Vangone, A.; Spinelli, R.; Scarano, V.; Cavallo, L.; Oliva, R. COCOMAPS: A web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics 2011, 27, 2915–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutsias, E.A.; Seok, C.; Jacobson, M.P.; Dill, K.A. A kinematic view of loop closure. J. Comput. Chem. 2004, 25, 510–528. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—a New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Abdel-Azeim, S.; Chermak, E.; Vangone, A.; Oliva, R.; Cavallo, L. MDcons: Intermolecular contact maps as a tool to analyze the interface of protein complexes from molecular dynamics trajectories. BMC Bioinform. 2014, 15, S1. [Google Scholar] [CrossRef] [Green Version]
- Oliva, R.; Chermak, E.; Cavallo, L. Analysis and Ranking of Protein-Protein Docking Models Using Inter-Residue Contacts and Inter-Molecular Contact Maps. Molecules 2015, 20, 12045–12060. [Google Scholar] [CrossRef]
- Vangone, A.; Cavallo, L.; Oliva, R. Using a consensus approach based on the conservation of inter-residue contacts to rank CAPRI models. Proteins 2013, 81, 2210–2220. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # S Protein Sequences | S929T | D936Y | S939F |
|---|---|---|---|
| 415,673 | 467 | 1296 | 1108 |
| Amino Acid | Pre-Fusion | Post-Fusion |
|---|---|---|
| T929 | exposed | partly buried (18.6%) a |
| Y936 | exposed | partly buried (19.0%) |
| F939 | exposed | exposed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliva, R.; Shaikh, A.R.; Petta, A.; Vangone, A.; Cavallo, L. D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect. Molecules 2021, 26, 2622. https://doi.org/10.3390/molecules26092622
Oliva R, Shaikh AR, Petta A, Vangone A, Cavallo L. D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect. Molecules. 2021; 26(9):2622. https://doi.org/10.3390/molecules26092622
Chicago/Turabian StyleOliva, Romina, Abdul Rajjak Shaikh, Andrea Petta, Anna Vangone, and Luigi Cavallo. 2021. "D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect" Molecules 26, no. 9: 2622. https://doi.org/10.3390/molecules26092622
APA StyleOliva, R., Shaikh, A. R., Petta, A., Vangone, A., & Cavallo, L. (2021). D936Y and Other Mutations in the Fusion Core of the SARS-CoV-2 Spike Protein Heptad Repeat 1: Frequency, Geographical Distribution, and Structural Effect. Molecules, 26(9), 2622. https://doi.org/10.3390/molecules26092622