
Sesquiterpene Lactones with Dual Inhibitory Activity against the Trypanosoma brucei Pteridine Reductase 1 and Dihydrofolate Reductase


 , and
, and 
Abstract
:1. Introduction
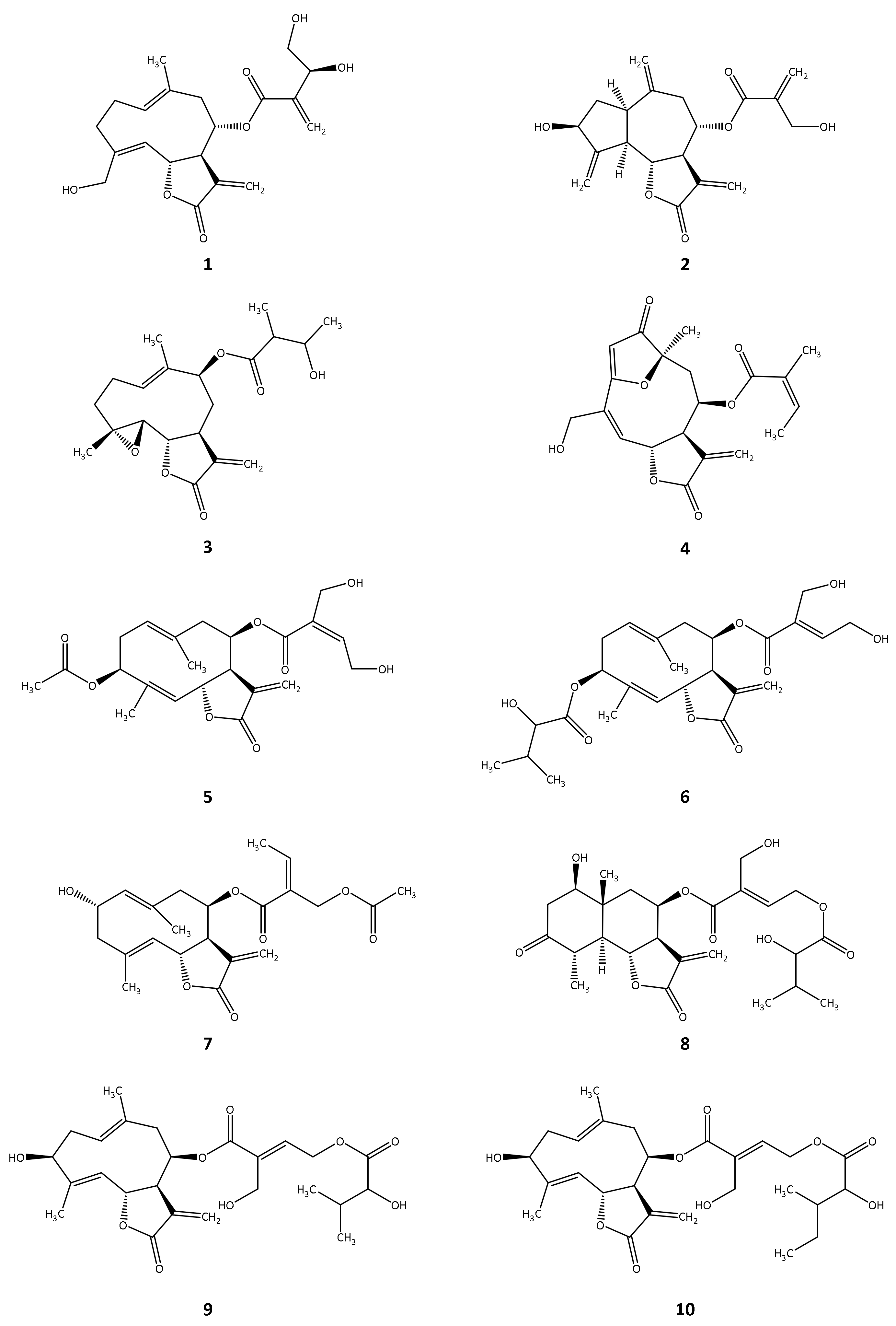
2. Results
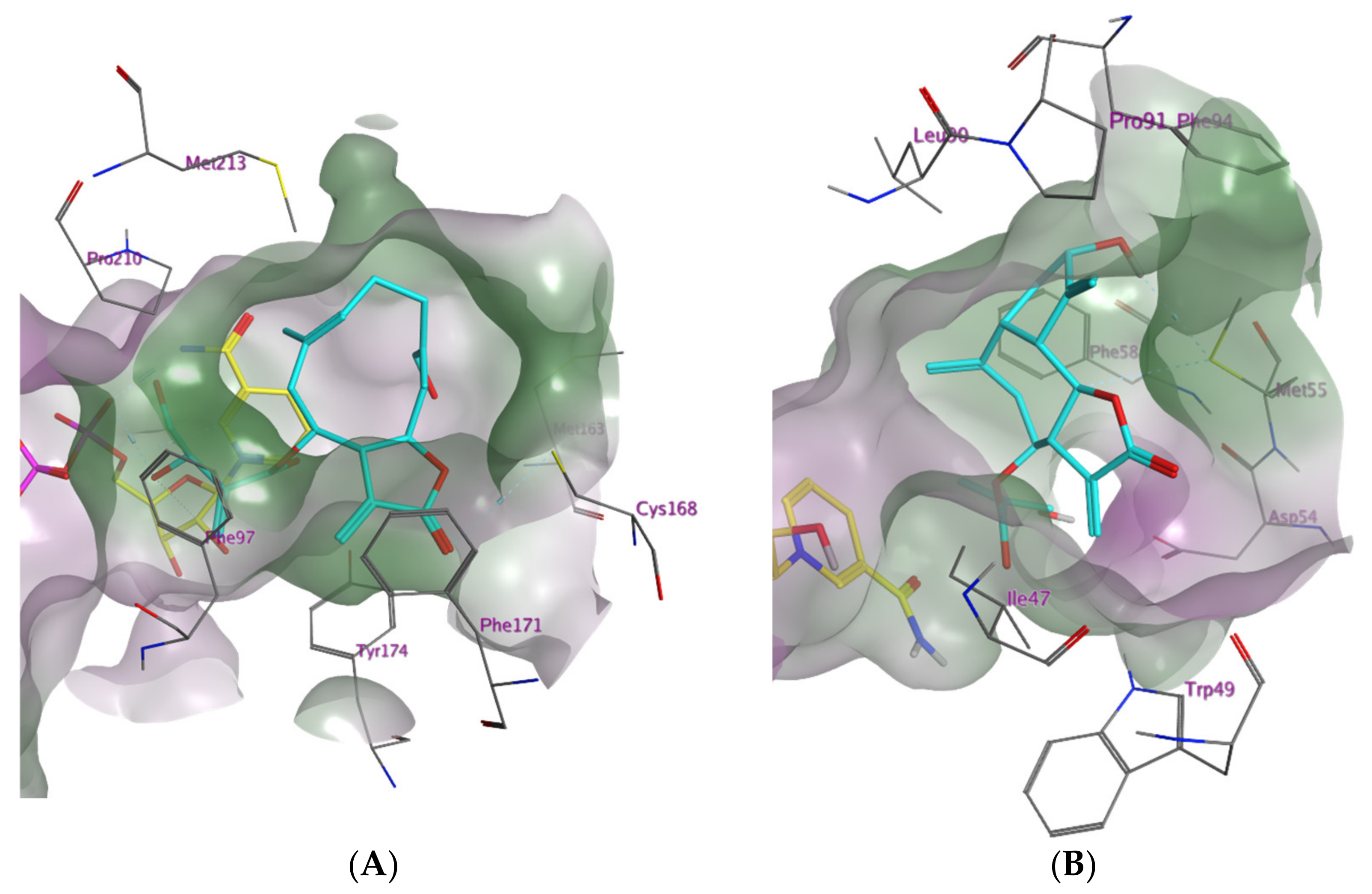
2.1. In Silico Investigation of Natural Products as Potential Inhibitors of TbPTR1 and TbDHFR
2.2. In Vitro Evaluation of the In Silico Hits against TbDHFR and TbPTR1
3. Discussion
4. Materials and Methods
4.1. In Silico Procedure
4.1.1. Preparation of the Respective 3D Protein Structures
4.1.2. Natural Product Databases
4.1.3. Pharmacophore Design
4.1.4. Virtual Screening
4.1.5. Docking Simulations
4.2. Recombinant Expression and Purification of TbPTR1
4.3. Cloning of TbDHFR into E. coli BL21(DE3) Host Strain
4.4. Recombinant Expression and Purification of TbDHFR
4.5. Kinetic Characterization of TbPTR1
4.6. Kinetic Characterization of TbDHFR
4.7. Test Compounds
4.8. Single Concentration Assays
4.9. Determination of the IC50 Values
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Neglected Tropical Diseases-Global. Available online: https://www.who.int/health-topics/neglected-tropical-diseases#tab=tab_1 (accessed on 22 September 2021).
- Neglected Tropical Diseases. Available online: https://www.who.int/health-topics/neglected-tropical-diseases#tab=tab_2 (accessed on 22 December 2021).
- Mitra, A.K.; Mawson, A.R. Neglected Tropical Diseases: Epidemiology and Global Burden. Trop. Med. Infect. Dis. 2017, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Trypanosomiasis, Human African (Sleeping Sickness). Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 8 September 2021).
- Deeks, E.D. Fexinidazole: First Global Approval. Drugs 2019, 79, 215–220. [Google Scholar] [CrossRef]
- Bonnet, J.; Boudot, C.; Courtioux, B. Overview of the Diagnostic Methods Used in the Field for Human African Trypanosomiasis: What Could Change in the Next Years? BioMed Res. Int. 2015, 2015, 583262. [Google Scholar] [CrossRef] [Green Version]
- Camara, M.; Ouattara, E.; Duvignaud, A.; Migliani, R.; Camara, O.; Leno, M.; Solano, P.; Bucheton, B.; Camara, M.; Malvy, D. Impact of the Ebola outbreak on Trypanosoma brucei gambiense infection medical activities in coastal Guinea, 2014–2015: A retrospective analysis from the Guinean national Human African Trypanosomiasis control program. PLoS Negl. Trop. Dis. 2017, 11, e0006060. [Google Scholar] [CrossRef]
- Relman, D.A.; Choffnes, E.R. The causes and Impacts of Neglected Tropical and Zoonotic Diseases: Opportunities for Integrated Intervention Strategies, Workshop Summary; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Lucius, R.; Loos-Frank, B.; Lane, R.P. Biologie Von Parasiten, 3rd ed.; Springer Spektrum: Heidelberg, Germany, 2018. [Google Scholar]
- Franco, J.R.; Simarro, P.P.; Diarra, A.; Jannin, J.G. Epidemiology of human African trypanosomiasis. CLEP 2014, 6, 257–275. [Google Scholar]
- Ong, H.B.; Sienkiewicz, N.; Wyllie, S.; Fairlamb, A.H. Dissecting the metabolic roles of pteridine reductase 1 in Trypanosoma brucei and Leishmania major. J. Biol. Chem. 2011, 286, 10429–10438. [Google Scholar] [CrossRef] [Green Version]
- Dewar, S.; Sienkiewicz, N.; Ong, H.B.; Wall, R.J.; Horn, D.; Fairlamb, A.H. The Role of Folate Transport in Antifolate Drug Action in Trypanosoma brucei. J. Biol. Chem. 2016, 291, 24768–24778. [Google Scholar] [CrossRef] [Green Version]
- Knighton, D.R.; Kan, C.C.; Howland, E.; Janson, C.A.; Hostomska, Z.; Welsh, K.M.; Matthews, D.A. Structure of and kinetic channelling in bifunctional dihydrofolate reductase-thymidylate synthase. Nat. Struct. Biol. 1994, 1, 186–194. [Google Scholar] [CrossRef]
- Gibson, M.W.; Dewar, S.; Ong, H.B.; Sienkiewicz, N.; Fairlamb, A.H. Trypanosoma brucei DHFR-TS Revisited: Characterisation of a Bifunctional and Highly Unstable Recombinant Dihydrofolate Reductase-Thymidylate Synthase. PLoS Negl. Trop. Dis. 2016, 10, e0004714. [Google Scholar] [CrossRef] [Green Version]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosomal dihydrofolate reductase reveals natural antifolate resistance. ACS Chem. Biol. 2011, 6, 905–911. [Google Scholar] [CrossRef]
- Nare, B.; Hardy, L.W.; Beverley, S.M. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J. Biol. Chem. 1997, 272, 13883–13891. [Google Scholar] [CrossRef] [Green Version]
- Kimuda, M.P.; Laming, D.; Hoppe, H.C.; Tastan Bishop, Ö. Identification of Novel Potential Inhibitors of Pteridine Reductase 1 in Trypanosoma brucei via Computational Structure-Based Approaches and in Vitro Inhibition Assays. Molecules 2019, 24, 142. [Google Scholar] [CrossRef] [Green Version]
- Adegboye, O.; Field, M.A.; Kupz, A.; Pai, S.; Sharma, D.; Smout, M.J.; Wangchuk, P.; Wong, Y.; Loiseau, C. Natural-Product-Based Solutions for Tropical Infectious Diseases. Clin. Microbiol. Rev. 2021, 34, e0034820. [Google Scholar] [CrossRef]
- Moujir, L.; Callies, O.; Sousa, P.M.C.; Sharopov, F.; Seca, A.M.L. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Appl. Sci. 2020, 10, 3001. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Da Costa, F.B.; Lopes, N.P.; Kaiser, M.; Brun, R. In Silico prediction and experimental evaluation of furanoheliangolide sesquiterpene lactones as potent agents against Trypanosoma brucei rhodesiense. Antimicrob. Agents Chemother. 2014, 58, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.J.; Nour, A.M.M.; Khalid, S.A.; Kaiser, M.; Brun, R. Quantitative structure--antiprotozoal activity relationships of sesquiterpene lactones. Molecules 2009, 14, 2062–2076. [Google Scholar] [CrossRef]
- Herrmann, F.C. In Silico-Identifikation Und in Vitro-Evaluation Natürlicher Inhibitoren Diverser Therapierelevanter Zielenzyme Von Humanpathogenen Eukaryoten der Gattungen Trypanosoma, Leishmania und Plasmodium. Ph.D. Thesis, Westfälische Wilhelms-Universität, Münster, Germany, 2016. [Google Scholar]
- Herrmann, F.C.; Sivakumar, N.; Jose, J.; Costi, M.P.; Pozzi, C.; Schmidt, T.J. In Silico Identification and In Vitro Evaluation of Natural Inhibitors of Leishmania major Pteridine Reductase, I. Molecules 2017, 22, 2166. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Mizuno, H.; Usuki, T. Ionic Liquid-Assisted Extraction and Isolation of Cynaropicrin and Cnicin from Artichoke and Blessed thistle. ChemistrySelect 2018, 3, 1781–1786. [Google Scholar] [CrossRef]
- Gökbulut, A.; Kaiser, M.; Brun, R.; Sarer, E.; Schmidt, T.J. 9β-hydroxyparthenolide esters from Inula montbretiana and their antiprotozoal activity. Planta Med. 2012, 78, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Kimani, N.M.; Matasyoh, J.C.; Kaiser, M.; Brun, R.; Schmidt, T.J. Antiprotozoal Sesquiterpene Lactones and Other Constituents from Tarchonanthus camphoratus and Schkuhria pinnata. J. Nat. Prod. 2018, 81, 124–130. [Google Scholar] [CrossRef]
- Nogueira Da Silva, M. The Use of Chemometric and Chemoinformatic Tools for Identification and Targeted Isolation of Compounds from Asteraceae with Antiprotozoal Activity. Ph.D. Thesis, Westfälische Wilhelms-Universität, Münster, Germany, 2016. [Google Scholar]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.; Biavatti, M.W.; Brun, R.; Da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases-part II. Curr. Med. Chem. 2012, 19, 2176–2228. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.; Biavatti, M.W.; Brun, R.; Da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases-part I. Curr. Med. Chem. 2012, 19, 2128–2175. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Brun, R.; Willuhn, G.; Khalid, S.A. Anti-trypanosomal activity of helenalin and some structurally related sesquiterpene lactones. Planta Med. 2002, 68, 750–751. [Google Scholar] [CrossRef]
- Trossini, G.H.G.; Maltarollo, V.G.; Schmidt, T.J. Hologram QSAR studies of antiprotozoal activities of sesquiterpene lactones. Molecules 2014, 19, 10546–10562. [Google Scholar] [CrossRef] [Green Version]
- Kimani, N.M.; Matasyoh, J.C.; Kaiser, M.; Nogueira, M.S.; Trossini, G.H.G.; Schmidt, T.J. Complementary Quantitative Structure—Activity Relationship Models for the Antitrypanosomal Activity of Sesquiterpene Lactones. Int. J. Mol. Sci. 2018, 19, 3721. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, T.J. Toxic activities of sesquiterpene lactones: Structural and biochemical aspects. Curr. Org. Chem. 1999, 3, 577–608. [Google Scholar]
- Zimmermann, S.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Cynaropicrin: The first plant natural product with in vivo activity against Trypanosoma brucei. Planta Med. 2012, 78, 553–556. [Google Scholar] [CrossRef]
- Zimmermann, S.; Oufir, M.; Leroux, A.; Krauth-Siegel, R.L.; Becker, K.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Cynaropicrin targets the trypanothione redox system in Trypanosoma brucei. Bioorg. Med. Chem. 2013, 21, 7202–7209. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- RCSB Protein Data Bank: Homepage. Available online: https://www.rcsb.org/ (accessed on 1 September 2021).
- Dawson, A.; Tulloch, L.B.; Barrack, K.L.; Hunter, W.N. High resolution structure of TbPTR1 in complex with Pemetrexed. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 1334. [Google Scholar] [CrossRef]
- Dawson, A.; Tulloch, L.B.; Barrack, K.L.; Hunter, W.N. Structure of PTR1 from Trypanosoma brucei in ternary complex with 2,4-diamino-5-[2-(2,5-dimethoxyphenyl)ethyl]thieno[2,3-d]-pyrimidine and NADP+. Acta Crystallogr. Sect. D Struct. Biol. 2010, 66, 1334–1340. [Google Scholar] [CrossRef]
- Khalaf, A.I.; Huggan, J.K.; Suckling, C.J.; Gibson, C.L.; Stewart, K.; Giordani, F.; Barrett, M.P.; Wong, P.E.; Barrack, K.L.; Hunter, W.N. Crystal structure of pteridine reductase 1 (PTR1) from Trypanosoma brucei in ternary complex with cofactor and inhibitor. J. Med. Chem. 2014, 57, 6479. [Google Scholar] [CrossRef] [Green Version]
- Landi, G.; Pozzi, C.; Di Pisa, F.; Dello lacono, L.; Mangani, S. Trypanosoma brucei PTR1 in complex with cofactor and inhibitor NMT-H024 (compound 2). J. Med. Chem. 2016, 59, 7598–7616. [Google Scholar] [CrossRef]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosoma brucei dihydrofolate reductase pyrimethamine complex. ACS Chem. Biol. 2011, 6. [Google Scholar] [CrossRef]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosoma brucei dihydrofolate reductase (TbDHFR) in complex with WR99210. ACS Chem. Biol. 2011, 6. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Tubeleviciute, A.; Teese, M.G.; Jose, J. Escherichia coli kduD encodes an oxidoreductase that converts both sugar and steroid substrates. Appl. Microbiol. Biotechnol. 2014, 98, 5471–5485. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Compound | TbPTR1 | TbDHFR | T. brucei rhodesiense | |||
|---|---|---|---|---|---|---|
| Inhibition at 100 µM (%) | IC50 (µM) | Inhibition at 50 µM (%) | IC50 (µM) | IC50 (µM) | Reference | |
| 1 | 63.9 | 21.2 a | 15.2 | 0.4 | [26] | |
| 2 | 92.1 | 12.4 | 95.8 | 7.1 | 0.3 | [26] |
| 3 | 73.1 | 40.5 | 63.8 | 13.3 | 1.3 | [27] |
| 4 | 79.9 | 30.5 | 34.9 | 0.1 | [21] | |
| 5 | 58.1 | 31.5 a | n.i. | 1.7 | [28] | |
| 6 | 33.1 | n.i. | 0.8 | [28] | ||
| 7 | 25.6 | 48.7 | 1.6 | [29] | ||
| 8 | 14.2 | 40.7 | 2.7 | [28] | ||
| 9 | 28.5 | 95.9 | n.d. | 2.6 | [28] | |
| 10 | 26.7 | 20.1 | 4.0 | [28] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Possart, K.; Herrmann, F.C.; Jose, J.; Costi, M.P.; Schmidt, T.J. Sesquiterpene Lactones with Dual Inhibitory Activity against the Trypanosoma brucei Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules 2022, 27, 149. https://doi.org/10.3390/molecules27010149
Possart K, Herrmann FC, Jose J, Costi MP, Schmidt TJ. Sesquiterpene Lactones with Dual Inhibitory Activity against the Trypanosoma brucei Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules. 2022; 27(1):149. https://doi.org/10.3390/molecules27010149
Chicago/Turabian StylePossart, Katharina, Fabian C. Herrmann, Joachim Jose, Maria P. Costi, and Thomas J. Schmidt. 2022. "Sesquiterpene Lactones with Dual Inhibitory Activity against the Trypanosoma brucei Pteridine Reductase 1 and Dihydrofolate Reductase" Molecules 27, no. 1: 149. https://doi.org/10.3390/molecules27010149
APA StylePossart, K., Herrmann, F. C., Jose, J., Costi, M. P., & Schmidt, T. J. (2022). Sesquiterpene Lactones with Dual Inhibitory Activity against the Trypanosoma brucei Pteridine Reductase 1 and Dihydrofolate Reductase. Molecules, 27(1), 149. https://doi.org/10.3390/molecules27010149