
Design, Synthesis, and Biological Evaluation of 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines as Microtubule Targeting Agents

 and
and
Abstract
:
1. Introduction
2. Results and Discussion
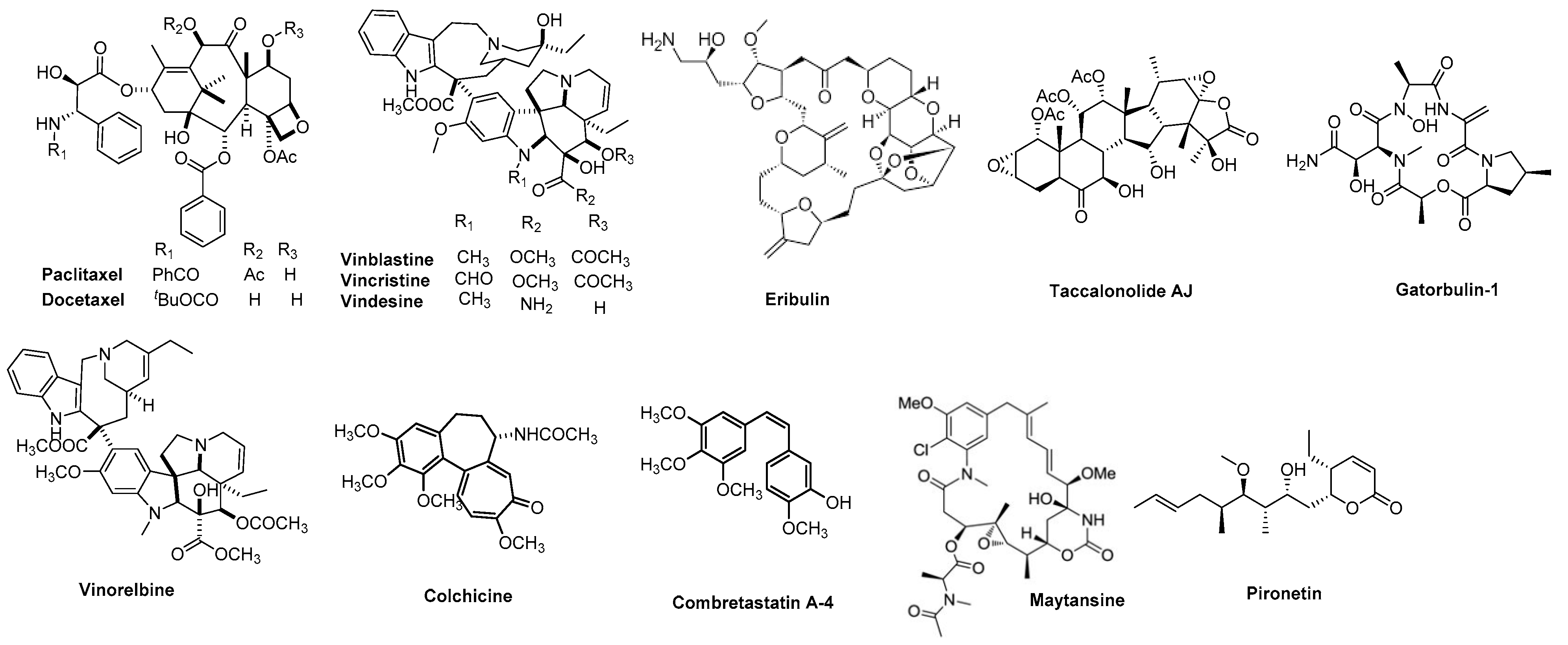
2.1. Rationale
- (1)
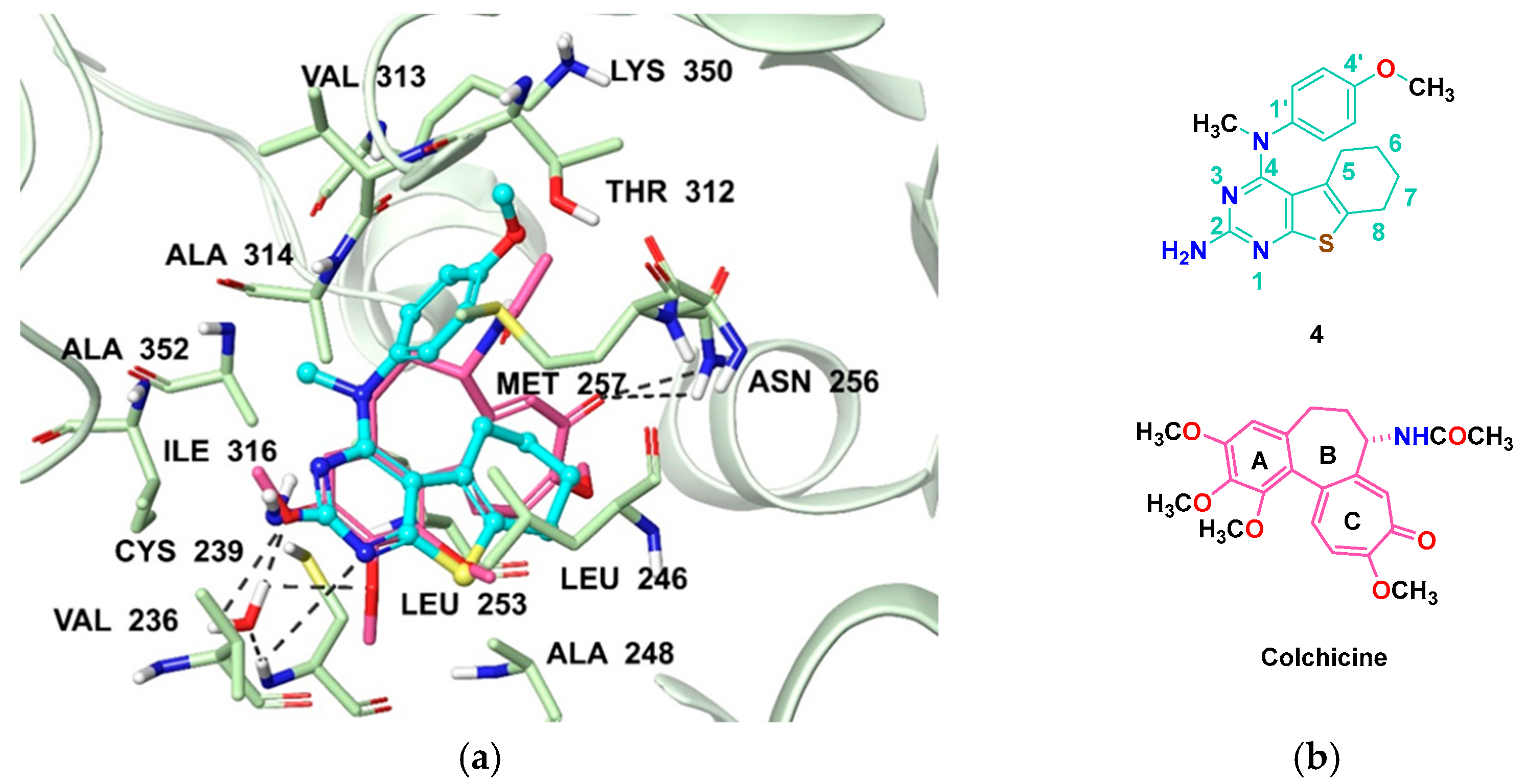
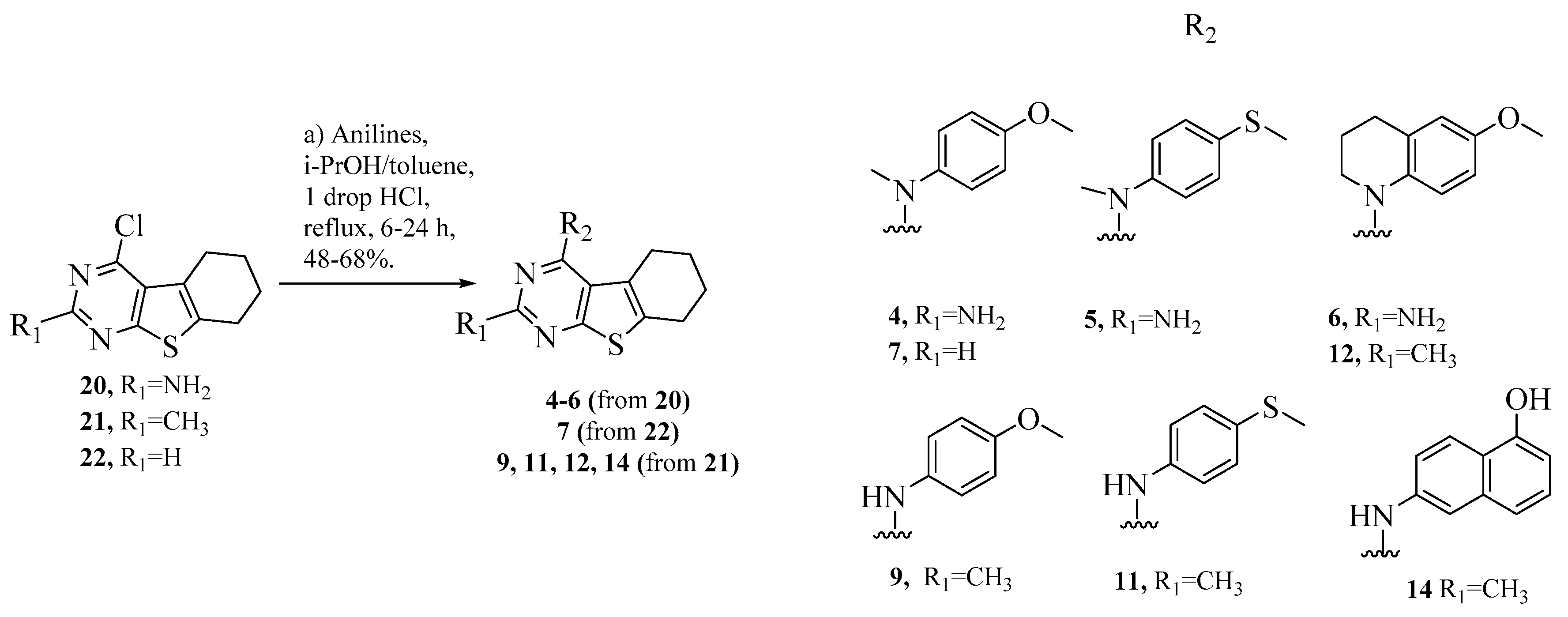
- Isosteric replacement: To explore the activities of compounds with the 4,5,6,7-tetrahydrobenzo thiophene scaffold on both inhibition of cancer cell proliferation and microtubule depolymerization, we carried out the isosteric replacement of the scaffold -NH- of the lead compounds 1–3 by sulfur (-S-) to afford target compounds 4–14 (Table 1). Isosteric replacement of -NH with (-S-) has literature precedence in improving antiproliferative and microtubule depolymerizing activities [27]. Moreover, pharmacological applications of 5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines have been extensively illustrated in various reports in the literature [28,29,30,31,32,33,34,35,36,37,38]. In addition, the lead tricyclic compounds and the proposed target compounds incorporate a p-methoxyphenyl substitution akin to colchicine and CA-4 (Figure 1). The nature of the heteroatom substitution (S for NH) affects hydrogen bond (HB) strength [39]. Thus, it was also of interest to isosterically replace the oxygen atom of the 4′-OCH3 of 4, 8 and 9 with a sulfur moiety to afford 5, 10, and 11, in analogy to 2.
- (2)
- Decrease numbers of sp2 bonds: Drug candidates show a higher clinical success rate with one or more sp3 hybridized carbon atoms as compared to “flat” molecules, due to low aqueous solubility of purely aromatic compounds [40]. One of the major limitation of some MTAs, particularly the taxanes, is their poor water solubility [41]. Thus, water-soluble MTAs are highly coveted, and an enormous effort continues to chemically modify and/or formulate analogues to increase their water solubility. Increasing ‘aromatic proportion’ in a molecule has a detrimental effect on the solubility [40]. The fraction of sp3 hybridized carbon atoms (Fsp3), in other words, the fraction of carbon atoms that are saturated, correlates positively with water solubility [40]. In an attempt to both increase the water solubility as well to probe the potential interactions with the hydrophobic pocket in the CS, we designed target compounds 4–14 by incorporating sp3 hybridized carbon atoms in the tricyclic scaffold of the lead compounds 1–3.
- (3)
- Variation of the substituents at the 2-position: Compound 7 was specifically designed to determine the effect of replacing the 2-NH2 in 4 with a 2-H. This allows an exploration of the 2-NH2 and hydrogen bond interactions with corresponding amino acids at the CS. It was also of our interest to observe the effect of isosteric replacement of 2-NH2 on compound 4 with a 2-CH3 to afford 8. This would also provide information regarding the activity on the replacement of H with CH3 at the 2-position in the tricyclic scaffold.
- (4)
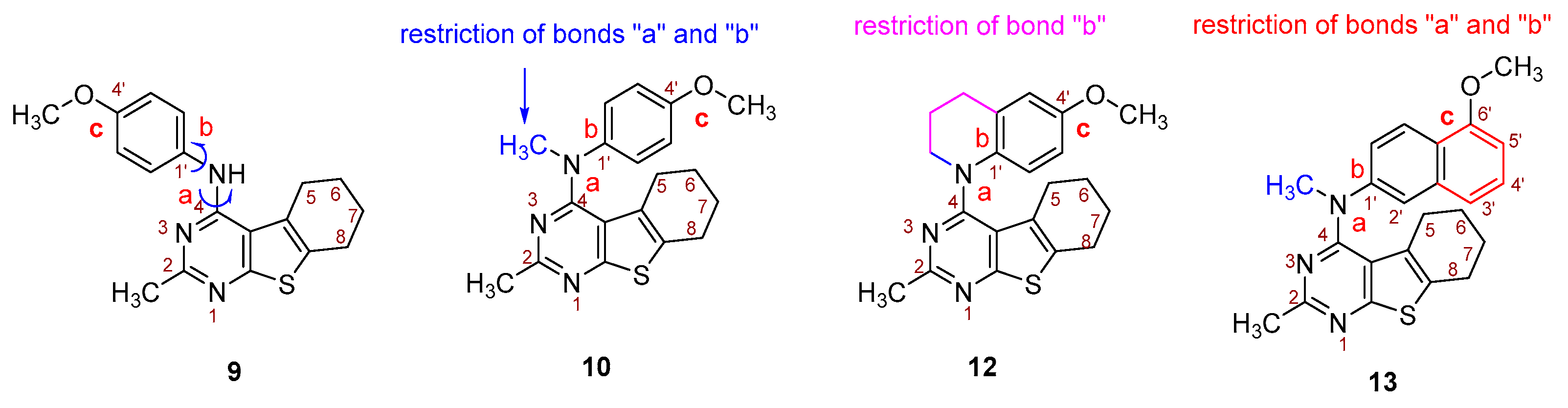
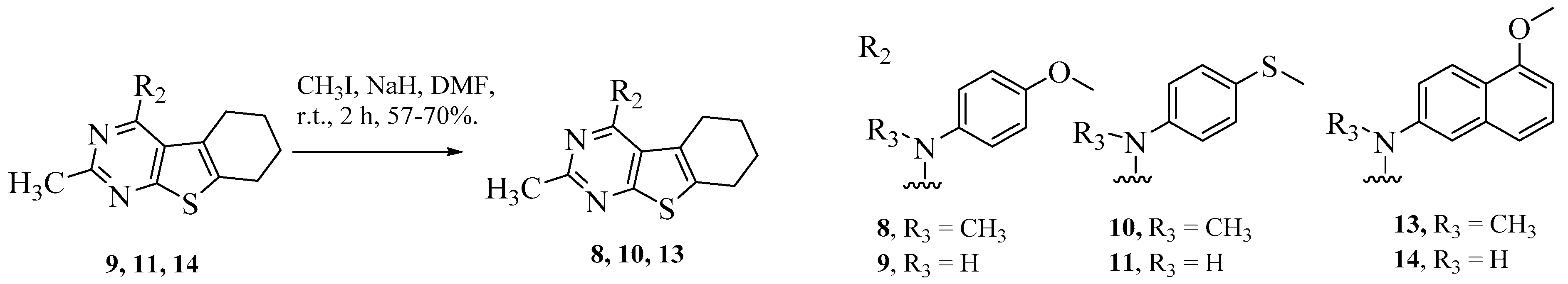
- Conformational restriction: Conformational restriction or rigidification of a ligand can decrease the entropic penalty [42]. The ligand can adopt a preferred conformation for binding, which might lead to enhanced potency for a given physiological target [42]. In an effort to better define the conformational requirements for biological activities, we systematically incorporated various groups to restrict bond rotations. The conformation of 9 (Figure 3) is determined by three rotatable single bonds: the 4-position C-N bond (bond a), the 1′-position C-N bond (bond b) and the 4′-position C-O bond (bond c). Conformational analysis via molecular modeling and 1H NMR studies [25] suggest that the methyl group on the aniline nitrogen in 1 restricted the free rotation of bond a as well as bond b (Figure 2) and consequently restricted the conformation of the anilino ring. To study the significance of conformational restriction on biological activities, we first designed compounds 8 and 9. In 9, the rotation of bonds a and b was restricted by incorporating a methyl group at the N4- position to afford compound 8. Incorporation of tetrahydroquinoline rings in 6 and 12 further restricted bond b of 4 and 8. The design of compound 13 via the incorporation of a 5-methoxy naphthalene ring provided a further element of conformational restriction.
2.2. Molecular Modeling
2.3. Chemistry
2.4. Biological Evaluations and Discussion
2.4.1. Antiproliferative and Microtubule Depolymerization Effects
2.4.2. Inhibition of Tubulin Assembly and Colchicine Binding
2.4.3. Effect on βIII-Tubulin and Pgp-Mediated Cancer Cell Resistance
2.4.4. Activity of Compound 4 in the NCI Cancer Cell Line Panel
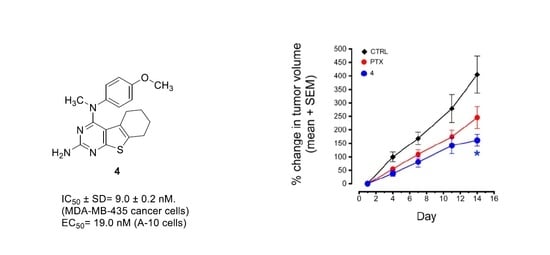
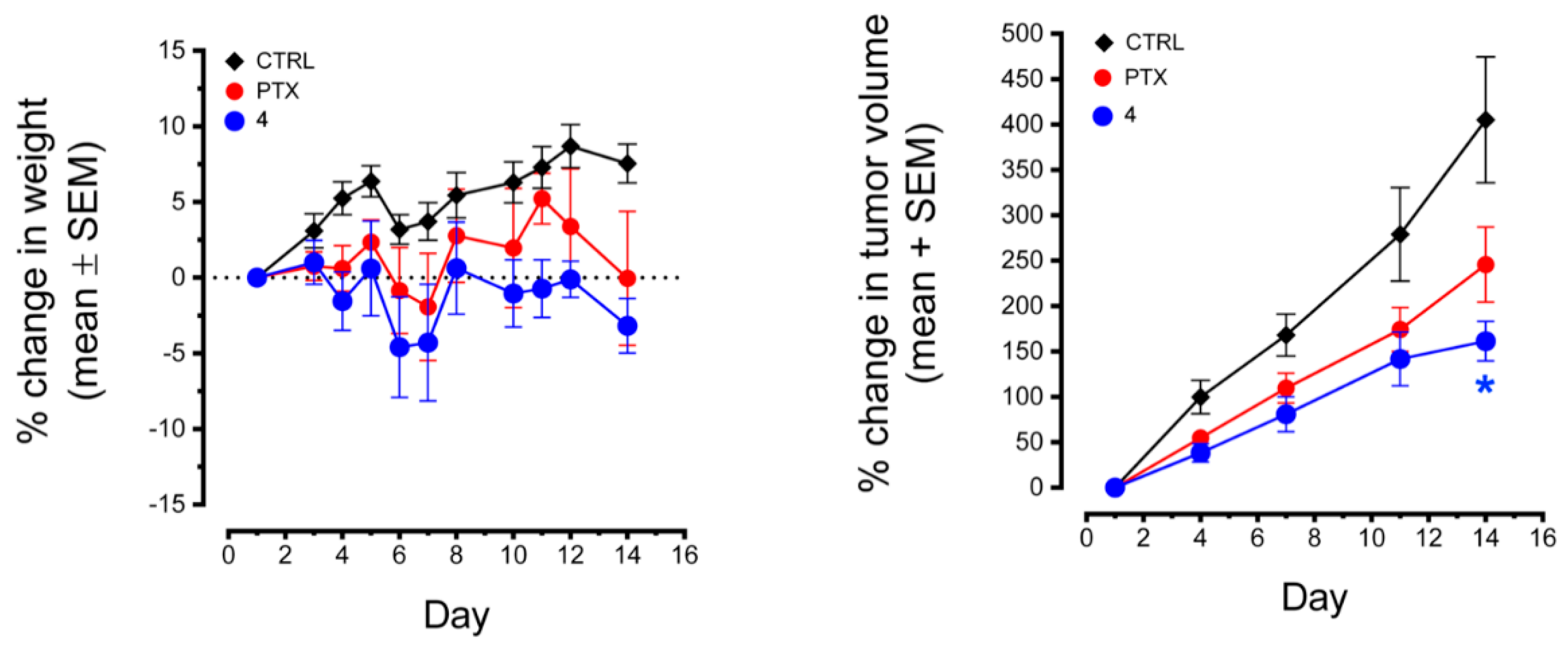
2.4.5. Antitumor Activity of Compound 4 in MDA-MB-435 Xenografts
3. Materials and Methods
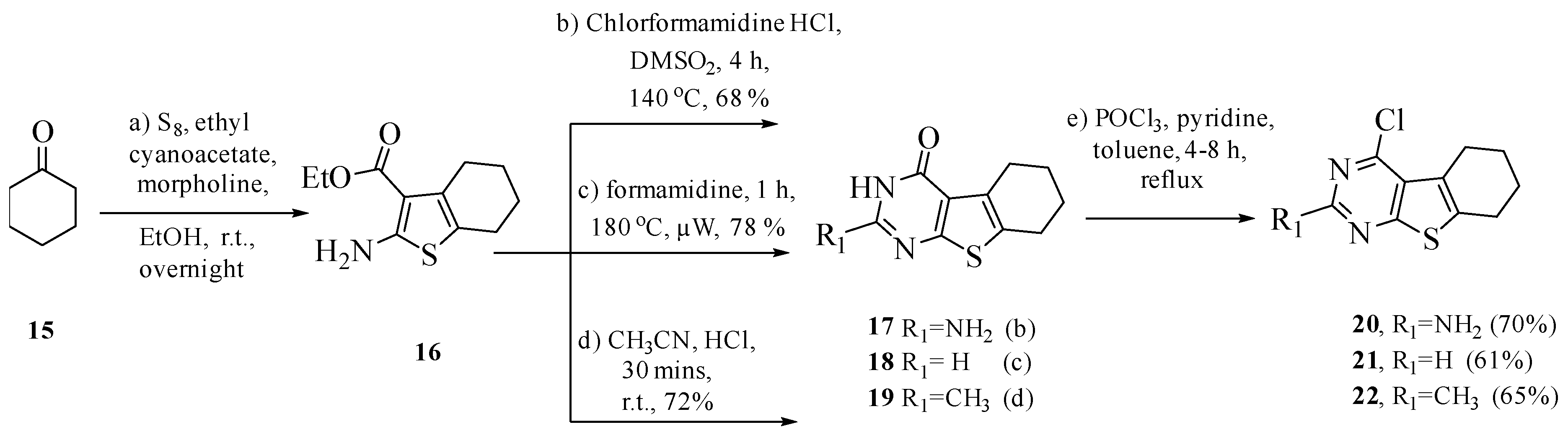
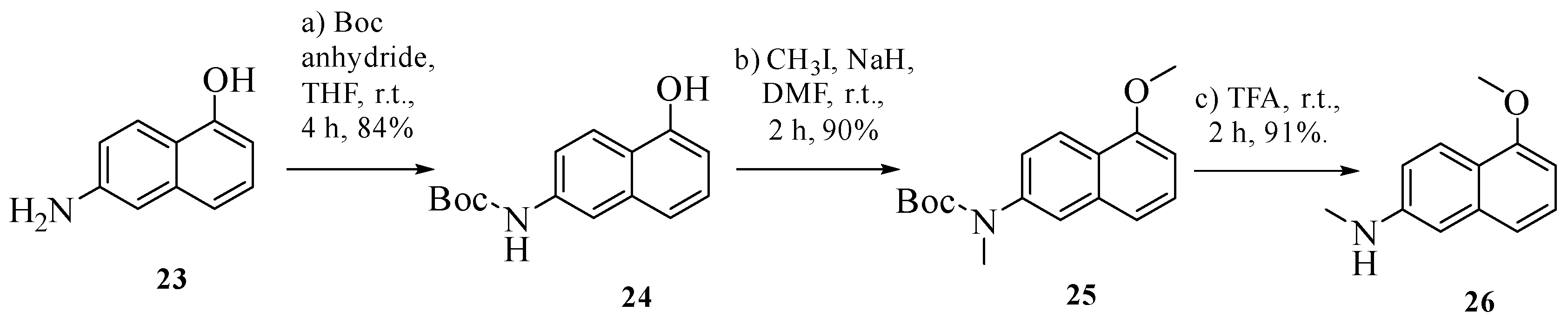
3.1. Chemistry
General Procedure for Synthesis of 4–14
3.2. Molecular Modeling
3.3. Biological Studies
3.3.1. Effects of Compounds on Cellular Microtubules
3.3.2. Sulforhodamine B (SRB) Assay
3.3.3. Quantitative Tubulin Studies
3.3.4. Cell Culture
3.3.5. MDA-MB-435 Xenograft Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–625. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [Green Version]
- Field, J.J.; Kanakkanthara, A.; Miller, J.H. Microtubule-targeting agents are clinically successful due to both mitotic and interphase impairment of microtubule function. Bioorganic Med. Chem. 2014, 22, 5050–5059. [Google Scholar] [CrossRef]
- de Forges, H.; Bouissou, A.; Perez, F. Interplay between microtubule dynamics and intracellular organization. Int. J. Biochem. Cell Biol. 2012, 44, 266–274. [Google Scholar] [CrossRef]
- Kaul, R.; Risinger, A.L.; Mooberry, S.L. Microtubule-Targeting Drugs: More than Antimitotics. J. Nat. Prod. 2019, 82, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, M.O.; Prota, A.E. Microtubule-Targeting Agents: Strategies to Hijack the Cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Matthew, S.; Chen, Q.-Y.; Ratnayake, R.; Fermaintt, C.S.; Lucena-Agell, D.; Bonato, F.; Prota, A.E.; Lim, S.T.; Wang, X.; Díaz, J.F.; et al. Gatorbulin-1, a distinct cyclodepsipeptide chemotype, targets a seventh tubulin pharmacological site. Proc. Natl. Acad. Sci. USA 2021, 118, e2021847118. [Google Scholar] [CrossRef] [PubMed]
- Rohena, C.C.; Mooberry, S.L. Recent progress with microtubule stabilizers: New compounds, binding modes and cellular activities. Nat. Prod. Rep. 2014, 31, 335–355. [Google Scholar] [CrossRef] [Green Version]
- Yee, S.S.; Du, L.; Risinger, A.L. Taccalonolide microtubule stabilizers. Prog. Chem. Org. Nat. Prod. 2020, 112, 183–206. [Google Scholar]
- Field, J.J.; Pera, B.; Calvo, E.; Canales, A.; Zurwerra, D.; Trigili, C.; Rodríguez-Salarichs, J.; Matesanz, R.; Kanakkanthara, A.; Wakefieldet, J.; et al. Zampanolide, a potent new microtubule-stabilizing agent, covalently reacts with the taxane luminal site in tubulin α,β-heterodimers and microtubules. Chem. Biol. 2012, 19, 686–698. [Google Scholar] [CrossRef]
- Liu, J.; Towle, M.J.; Cheng, H.; Saxton, P.; Reardon, C.; Wu, J.; Murphy, E.A.; Kuznetsov, G.; Johannes, C.W.; Tremblay, M.R.; et al. In vitro and in vivo anticancer activities of synthetic (-)-laulimalide, a marine natural product microtubule stabilizing agent. Anticancer Res. 2007, 27, 1509–1518. [Google Scholar]
- Kanakkanthara, A.; Northcote, P.T.; Miller, J.H. Peloruside A: A lead non-taxoid-site microtubule-stabilizing agent with potential activity against cancer, neurodegeneration, and autoimmune disease. Nat. Prod. Rep. 2016, 33, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Díaz, J.F.; Marsh, M.; Cuevas, C.; Liniger, M.; Neuhaus, C.; Andreu, J.M.; Altmann, K.-H.; Steinmetz, M.O. A new tubulin-binding site and pharmacophore for microtubule-destabilizing anticancer drugs. Proc. Natl. Acad. Sci. USA 2014, 111, 13817–13821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, A.E.; Setter, J.; Waight, A.B.; Bargsten, K.; Murga, J.; Díaz, J.F.; Steinmetz, M.O. Pironetin Binds Covalently to αCys316 and Perturbs a Major Loop and Helix of α-Tubulin to Inhibit Microtubule Formation. J. Mol. Biol. 2016, 428, 2981–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dybdal-Hargreaves, N.F.; Risinger, A.L.; Mooberry, S.L. Eribulin mesylate: Mechanism of action of a unique microtubule-targeting agent. Clin. Cancer Res. 2015, 21, 2445–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.A.; Shang, X.; Burlingame, S.M.; Okcu, M.F.; Ge, N.; Russell, H.V.; Egler, R.A.; David, R.D.; Vasudevan, S.A.; Yang, J.; et al. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Duran, G.E.; Wang, Y.C.; Francisco, E.B.; Rose, J.; Martinez, F.J.; Coller, J.; Brassard, D.; Vrignaud, P.; Sikic, B.I. Mechanisms of Resistance to Cabazitaxel. Mol. Cancer Ther. 2014, 14, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Li, B.; Fang, Y.; Liu, Y.; Wang, Y.; Li, J.; Zhou, W.; Wang, X. Overexpression of Class III β-tubulin, Sox2, and nuclear Survivin is predictive of taxane resistance in patients with stage III ovarian epithelial cancer. BMC Cancer 2015, 15, 536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.-T.; Liu, Y.-N.; Liu, Z.-P. Tubulin colchicine binding site inhibitors as vascular disrupting agents in clinical developments. Curr. Med. Chem. 2015, 22, 1348–1360. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An Overview of Tubulin Inhibitors That Interact with the Colchicine Binding Site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, E.C.; O′Boyle, N.M. Colchicine-Binding Site Inhibitors from Chemistry to Clinic: A Review. Pharmaceuticals 2020, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Echteld, I.; Wechalekar, M.D.; Schlesinger, N.; Buchbinder, R.; Aletaha, D. Colchicine for acute gout. Cochrane Database Syst. Rev. 2014, 15, CD006190. [Google Scholar] [CrossRef] [PubMed]
- Stengel, C.; Newman, S.P.; Leese, M.P.; Potter, B.V.; Reed, M.J.; Purohit, A. Class III beta-tubulin expression and in vitro resistance to microtubule targeting agents. Br. J. Cancer 2010, 102, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Devambatla, R.K.V.; Li, W.; Zaware, N.; Choudhary, S.; Hamel, E.; Mooberry, S.L.; Gangjee, A. Design, synthesis, and structure-activity relationships of pyrimido[4,5-b]indole-4-amines as microtubule depolymerizing agents that are effective against multidrug resistant cells. Bioorg. Med. Chem. Lett. 2017, 27, 3423–3430. [Google Scholar] [CrossRef]
- Gangjee, A.; Zaware, N.; Devambatla, R.K.V.; Raghavan, S.; Westbrook, C.D.; Dybdal-Hargreaves, N.F.; Hamel, E.; Mooberry, S.L. Synthesis of N4-(substituted phenyl)-N4-alkyl/desalkyl-9H-pyrimido[4,5-b]indole-2,4-diamines and identification of new microtubule disrupting compounds that are effective against multidrug resistant cells. Bioorg. Med. Chem. 2012, 21, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Choudhary, S.; Hamel, E.; Mooberry, S.L.; Gangjee, A. Structure based drug design and in vitro metabolism study: Discovery of N-(4-methylthiophenyl)-N,2-dimethyl-cyclopenta[d]pyrimidine as a potent microtubule targeting agent. Bioorg. Med. Chem. 2018, 26, 2437–2451. [Google Scholar] [CrossRef]
- Al-Taisan, K.M.; Al-Hazimi, H.M.A.; Al-Shihry, S.S. Synthesis, Characterization and Biological Studies of Some Novel Thieno[2,3-d]pyrimidines. Molecules 2010, 15, 3932–3957. [Google Scholar] [CrossRef]
- Aly, A.A.; Ishak, E.A.; Ramadan, M.; Germoush, M.O.; El-Emary, T.I.; Al-Muaikel, N.S. Recent Report on Thieno[2,3-d]pyrimidines. Their Preparation Including Microwave and Their Utilities in Fused Heterocycles Synthesis. J. Heterocycl. Chem. 2013, 50, 451–472. [Google Scholar] [CrossRef]
- Chitikina, S.S.; Buddiga, P.; Deb, P.K.; Mailavaram, R.P.; Venugopala, K.N.; Nair, A.B.; Al-Jaidi, B.; Kar, S. Synthesis and anthelmintic activity of some novel (E)-2-methyl/propyl-4-(2-(substitutedbenzylidene)hydrazinyl)-5,6,7,8-tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines. Med. Chem. Res. 2020, 29, 1600–1610. [Google Scholar] [CrossRef]
- Elmetwally, S.A.; Saied, K.F.; Eissa, I.H.; Elkaeed, E.B. Design, synthesis and anticancer evaluation of thieno[2,3-d]pyrimidine derivatives as dual EGFR/HER2 inhibitors and apoptosis inducers. Bioorg. Chem. 2019, 88, 102944. [Google Scholar] [CrossRef] [PubMed]
- Lao, C.; Zhou, X.; Chen, H.; Wei, F.; Huang, Z.; Bai, C. 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine derivatives as inhibitors of full-length RORγt. Bioorg. Chem. 2019, 90, 103077. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, A.; Prasad, T.; Sharma, T. Pyrimidine: A review on anticancer activity with key emphasis on SAR. Future J. Pharm. Sci. 2021, 7, 123. [Google Scholar] [CrossRef]
- Pal, K.; Raza, M.K.; Legac, J.; Ataur Rahman, M.; Manzoor, S.; Rosenthal, P.J.; Hoda, N. Design, synthesis, crystal structure and anti-plasmodial evaluation of tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine derivatives. RSC Med. Chem. 2021, 12, 970–981. [Google Scholar] [CrossRef]
- Pavase, L.S.; Mane, D.V. Synthesis and anticancer activities of novel (tetrahydrobenzo [4,5] thieno [2,3-d] pyrimidine-4-yl)-pyrolidine-2-carboxylic acid derivatives. Med. Chem. Res. 2016, 25, 2380–2391. [Google Scholar] [CrossRef]
- Pédeboscq, S.; Gravier, D.; Casadebaig, F.; Hou, G.; Gissot, A.; De Giorgi, F.; Ichas, F.; Cambar, J.; Pometan, J.-P. Synthesis and study of antiproliferative activity of novel thienopyrimidines on glioblastoma cells. Eur. J. Med. Chem. 2010, 45, 2473–2479. [Google Scholar] [CrossRef]
- Sharaky, M.; Kamel, M.; Aziz, M.A.; Omran, M.; Rageh, M.M.; Abouzid, K.A.M.; Shouman, S.A. Design, synthesis and biological evaluation of a new thieno[2,3-d]pyrimidine-based urea derivative with potential antitumor activity against tamoxifen sensitive and resistant breast cancer cell lines. J. Enzym. Inhib. Med. Chem. 2020, 35, 1641–1656. [Google Scholar] [CrossRef]
- Wei, F.; Zhou, X.; Chen, H.; Tian, X.; Liu, Z.; Yu, B.; He, X.; Bai, C.; Huang, Z. 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidine derivative attenuates lupus nephritis with less effect to thymocyte development. Immunol. Res. 2021, 69, 378–390. [Google Scholar] [CrossRef]
- Hao, M.-H. Theoretical Calculation of Hydrogen-Bonding Strength for Drug Molecules. J. Chem. Theory Comput. 2006, 2, 863–872. [Google Scholar] [CrossRef]
- Ritchie, T.J.; Macdonald, S.J.; Young, R.J.; Pickett, S.D. The impact of aromatic ring count on compound developability: Further insights by examining carbo- and hetero-aromatic and -aliphatic ring types. Drug Discov. Today 2011, 16, 164–171. [Google Scholar] [CrossRef]
- Britten, C.D.; Baker, S.D.; Denis, L.J.; Johnson, T.; Drengler, R.; Siu, L.L.; Duchin, K.; Kuhn, J.; Rowinsky, E.K. Oral paclitaxel and concurrent cyclosporin A: Targeting clinically relevant systemic exposure to paclitaxel. Clin. Cancer Res. 2000, 6, 3459–3468. [Google Scholar]
- Blundell, C.D.; Packer, M.J.; Almond, A. Quantification of free ligand conformational preferences by NMR and their relationship to the bioactive conformation. Bioorg. Med. Chem. 2013, 21, 4976–4987. [Google Scholar] [CrossRef]
- Banerjee, S.; Arnst, K.E.; Wang, Y.; Kumar, G.; Deng, S.; Yang, L.; Li, G.-B.; Yang, J.; White, W.S.; Li, W.; et al. Heterocyclic-Fused Pyrimidines as Novel Tubulin Polymerization Inhibitors Targeting the Colchicine Binding Site: Structural Basis and Antitumor Efficacy. J. Med. Chem. 2018, 61, 1704–1718. [Google Scholar] [CrossRef]
- Schrӧdinger. Release 2020-2: Maestro, Schrodinger; Schrӧdinger LLC: New York, NY, USA, 2020. [Google Scholar]
- Gangjee, A.; Pavana, R.K.; Ihnat, M.A.; Thorpe, J.E.; Disch, B.C.; Bastian, A.; Bailey-Downs, L.C.; Hamel, E.; Bai, R. Discovery of Antitubulin Agents with Antiangiogenic Activity as Single Entities with Multitarget Chemotherapy Potential. ACS Med. Chem. Lett. 2014, 5, 480–484. [Google Scholar] [CrossRef]
- Islam, F.; Quadery, T.M. Therapeutic potential, synthesis, patent evaluation and SAR studies of thieno[3,2-d]pyrimidine derivatives: Recent updates. Curr. Drug Targets 2021, 22, 1944–1963. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Quadery, T.M.; Bai, R.; Luckett-Chastain, L.R.; Hamel, E.; Ihnat, M.A.; Gangjee, A. Novel pyrazolo[4,3-d]pyrimidine microtubule targeting agents (MTAs): Synthesis, structure–activity relationship, in vitro and in vivo evaluation as antitumor agents. Bioorg. Med. Chem. Lett. 2021, 41, 127923. [Google Scholar] [CrossRef]
- Xiang, W.; Quadery, T.M.; Hamel, E.; Luckett-Chastain, L.R.; Ihnat, M.A.; Mooberry, S.L.; Gangjee, A. The 3-D conformational shape of N-naphthyl-cyclopenta[d]pyrimidines affects their potency as microtubule targeting agents and their antitumor activity. Bioorg. Med. Chem. 2020, 29, 115887. [Google Scholar] [CrossRef] [PubMed]
- Golani, L.; Islam, F.; O’Connor, C.; Dekhne, A.S.; Hou, Z.; Matherly, L.H.; Gangjee, A. Design, synthesis and biological evaluation of novel pyrrolo[2,3-d]pyrimidine as tumor-targeting agents with selectivity for tumor uptake by high affinity folate receptors over the reduced folate carrier. Bioorg. Med. Chem. 2020, 28, 115544. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E. Evaluation of antimitotic agents by quantitative comparisons of their effects on the polymerization of purified tubulin. Cell Biochem. Biophys. 2003, 38, 1–21. [Google Scholar] [CrossRef]
- Bogolubsky, A.V.; Ryabukhin, S.V.; Plaskon, A.S.; Stetsenko, S.V.; Volochnyuk, D.; Tolmachev, A.A. Dry HCl in Parallel Synthesis of Fused Pyrimidin-4-ones. J. Comb. Chem. 2008, 10, 858–862. [Google Scholar] [CrossRef]
- Lee, L.; Robb, L.M.; Lee, M.; Davis, R.; Mackay, H.; Chavda, S.; Babu, B.; O’Brien, E.L.; Risinger, A.L.; Mooberry, S.L.; et al. Design, Synthesis, and Biological Evaluations of 2,5-Diaryl-2,3-dihydro-1,3,4-oxadiazoline Analogs of Combretastatin-A4. J. Med. Chem. 2009, 53, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Risinger, A.L.; Jackson, E.M.; Polin, L.A.; Helms, G.L.; Leboeuf, D.A.; Joe, P.A.; Hopper-Borge, E.; Ludueña, R.F.; Kruh, G.D.; Mooberry, S.L. The Taccalonolides: Microtubule Stabilizers That Circumvent Clinically Relevant Taxane Resistance Mechanisms. Cancer Res. 2008, 68, 8881–8888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamel, E.; Lin, C.M. Separation of active tubulin and microtubule-associated proteins by ultracentrifugation and isolation of a component causing the formation of microtubule bundles. Biochemistry 1984, 23, 4173–4184. [Google Scholar] [CrossRef] [PubMed]
- Verdier-Pinard, P.; Lai, J.-Y.; Yoo, H.-D.; Yu, J.; Marquez, B.; Nagle, D.G.; Nambu, M.; White, J.D.; Falck, J.R.; Gerwick, W.H.; et al. Structure-Activity Analysis of the Interaction of Curacin A, the Potent Colchicine Site Antimitotic Agent, with Tubulin and Effects of Analogs on the Growth of MCF-7 Breast Cancer Cells. Mol. Pharmacol. 1998, 53, 62–76. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
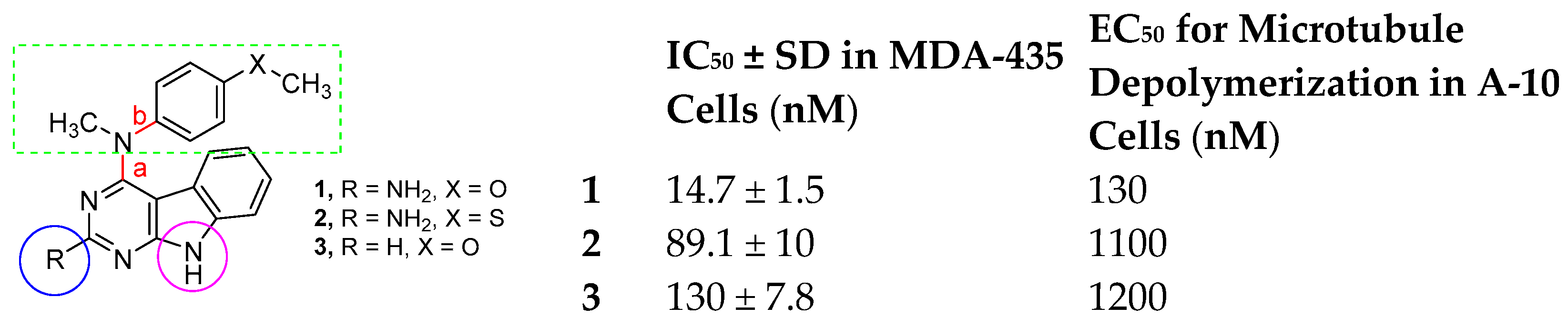{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound No. | R1 | R2 |
|---|---|---|
| 4 | NH2 |  |
| 5 | NH2 |  |
| 6 | NH2 |  |
| 7 | H |  |
| 8 | CH3 |  |
| 9 | CH3 |  |
| 10 | CH3 |  |
| 11 | CH3 |  |
| 12 | CH3 |  |
| 13 | CH3 |  |
| 14 | CH3 |  |
| Compound No | IC50 ± SD in MDA-435 Cells (nM) | EC50 for Microtubule Depolymerization in A-10 Cells (nM) | EC50/IC50 Ratio |
|---|---|---|---|
| 1a | 14.7 ± 1.5 | 130 | 8.8 |
| 2b | 89.1 ± 10 | 1100 | 12 |
| 3c | 130 ± 7.8 | 1200 | 9.2 |
| 4 | 9.0 ± 0.2 | 19 | 2.2 |
| 5 | 38.6 ± 5.6 | 70 | 1.8 |
| 6 | 59.6 ± 11.8 | 121 | 2.0 |
| 7 | 36.8 ± 5.2 | 45 | 1.2 |
| 8 | 53.0 ± 0.3 | 52 | 1.0 |
| 9 | ND | Not MT active c | ND |
| 10 | 87.7 ± 4.7 | 157 | 1.8 |
| 11 | ND | Not MT active c | ND |
| 12 | 125 ± 14 | 150 | 1.2 |
| 13 | 81.3 ± 8.4 | 118 | 1.5 |
| 14 | ND | Not MT active c | ND |
| CA-4 | 3.4 ± 0.6 | 13 | 3.8 |
| Compound | Inhibition of Tubulin Assembly IC50 ± SD (μM) | Inhibition of Colchicine Binding (% inhibition ± SD) at 5 μM |
|---|---|---|
| 1a | 1.4 ± 0.007 | 84 ± 0.50 |
| 2b | 2.3 ± 0.30 | 67.0 ± 5.0 |
| 3b | 2.3 ± 0.40 | 62.0 ± 4.0 |
| 4 | 0.82 ± 0.04 | 99.0 ± 1.0 |
| 5 | 0.49 ± 0.08 | 94.0 ± 2.0 |
| 7 | 0.49 ± 0.06 | 95.0 ± 0.40 |
| 8 | 0.56 ± 0.09 | 96.0 ± 0.40 |
| 10 | 0.54 ± 0.08 | 89.0 ± 0.20 |
| CA-4 | 1.0 ± 0.09 | 99.0 ± 0.20 |
| No. | IC50 ± SD in HeLa (nM) | IC50 ± SD in HeLa WTβ3 (nM) | Rr Value (WT β3/HeLa) | IC50 ± SD in SK-OV-3 Cells (nM) | IC50 ± SD in SK-OV-3 MDR1-M6/6 Cells (nM) | Rr Value (M6/6/SK-OV-3) |
|---|---|---|---|---|---|---|
| 1a | 21.3 ± 2.2 | 21.4 ± 3.5 | 1.0 | 27.6 ± 1.8 | 34.4 ± 5.9 | 1.2 |
| 2b | 118 ± 13 | 78.4 ± 4 | 0.7 | 156 ± 16 | 160 ± 15 | 1 |
| 3b | 142 ± 8.1 | 99.5 ± 12 | 0.8 | 173 ± 8.6 | 224 ± 21 | 1.4 |
| 4 | 13.5 ± 1.5 | 10.6 ± 1.8 | 0.8 | 11.8 ± 1.1 | 17.5 ± 0.8 | 1.5 |
| 5 | 64.3 ± 4.3 | 44.1 ± 4.8 | 0.7 | 71.8 ± 5.3 | 74.1 ± 11 | 1 |
| 6 | 123 ± 12 | 80.3 ± 6.4 | 0.7 | 92.5 ± 3.3 | 91.7 ± 12 | 1 |
| 7 | 57.2 ± 8.5 | 38.0 ± 3.9 | 0.7 | 37.0 ± 6.2 | 47.1 ± 7.6 | 1.3 |
| 8 | 87.9 ± 8.8 | 81.3 ± 5.2 | 0.9 | 47.7 ± 1.2 | 57.5 ± 0.5 | 1.2 |
| 10 | 146 ± 14 | 108 ± 8.6 | 0.7 | 136 ± 15 | 209 ± 40 | 1.5 |
| 12 | 191 ± 8.1 | 203 ± 12 | 1.1 | 178 ± 1.1 | 223 ± 9.4 | 1.3 |
| 13 | 170 ± 34 | 139 ± 20 | 0.8 | 142 ± 22 | 143 ± 244 | 1 |
| Paclitaxel | 2.8 ± 0.4 | 24.0 ± 3 | 8.6 | 5.0 ± 0.6 | 1200 ± 58 | 240 |
| CA-4 c | 3.3 ± 0.4 | 3.3 ± 0.3 | 1 | 5.5 ± 0.5 | 7.2 ± 1.1 | 1.3 |
| Panel/Cell Line | GI50 (nM) | Panel/Cell Line | GI50 (nM) | Panel/Cell Line | GI50 (nM) | Panel/Cell Line | GI50 (nM) |
|---|---|---|---|---|---|---|---|
| Leukemia | Colon Cancer | Melanoma | Renal Cancer | ||||
| CCRF-CEM | 7.12 | COLO 205 | 10.85 | LOX IMVI | 10.05 | 786-0 | 12.44 |
| HL-60(TB) | 4.38 | HCC-2998 | 11.78 | MALME-3M | 12.24 | A498 | 12.01 |
| K-562 | 2.72 | HCT-116 | 5.46 | M14 | 4.32 | ACHN | 10.23 |
| MOLT-4 | 8.80 | HCT-15 | 8.85 | MDA-MB-435 | 4.89 | CAKI-1 | 16.84 |
| RPMI-8226 | 12.67 | HT29 | 6.21 | SK-MEL-2 | 11.77 | RXF 393 | 7.65 |
| NSCLC | KM12 | 13.45 | SK-MEL-28 | 12.11 | SN12C | 14.36 | |
| A549/ATCC | 11.98 | SW-620 | 5.63 | SK-MEL-5 | 10.83 | TK-10 | 14.21 |
| EKVX | 11.24 | CNS Cancer | UACC-257 | 18.42 | UO-31 | 11.82 | |
| HOP-62 | 17.67 | SF-268 | 15.89 | UACC-62 | 14.88 | Breast Cancer | |
| HOP-92 | 14.05 | SF-295 | 10.23 | Ovarian Cancer | MCF7 | 8.01 | |
| NCI-H226 | 17.46 | SF-539 | 7.93 | IGROVI | 12.83 | MDA-MB-231/ATCC | 10.93 |
| NCI-H23 | 11.32 | SNB-19 | 9.04 | OVCAR-3 | 17.08 | HS 578T | 13.46 |
| NCI-H322M | 11.58 | SNB-75 | 8.42 | OVCAR-4 | 9.98 | BT-549 | 17.48 |
| NCI-H460 | 10.39 | U251 | 9.60 | OVCAR-5 | 10.78 | MDA-MB-468 | 6.05 |
| NCI-H522 | 6.87 | Prostate Cancer | OVCAR-8 | 18.10 | |||
| PC-3 | 7.67 | NCI/ADR-RES | 6.19 | ||||
| DU-145 | 15.64 | SK-OV-3 | 15.60 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, F.; Doshi, A.; Robles, A.J.; Quadery, T.M.; Zhang, X.; Zhou, X.; Hamel, E.; Mooberry, S.L.; Gangjee, A. Design, Synthesis, and Biological Evaluation of 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines as Microtubule Targeting Agents. Molecules 2022, 27, 321. https://doi.org/10.3390/molecules27010321
Islam F, Doshi A, Robles AJ, Quadery TM, Zhang X, Zhou X, Hamel E, Mooberry SL, Gangjee A. Design, Synthesis, and Biological Evaluation of 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines as Microtubule Targeting Agents. Molecules. 2022; 27(1):321. https://doi.org/10.3390/molecules27010321
Chicago/Turabian StyleIslam, Farhana, Arpit Doshi, Andrew J. Robles, Tasdique M. Quadery, Xin Zhang, Xilin Zhou, Ernest Hamel, Susan L. Mooberry, and Aleem Gangjee. 2022. "Design, Synthesis, and Biological Evaluation of 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines as Microtubule Targeting Agents" Molecules 27, no. 1: 321. https://doi.org/10.3390/molecules27010321
APA StyleIslam, F., Doshi, A., Robles, A. J., Quadery, T. M., Zhang, X., Zhou, X., Hamel, E., Mooberry, S. L., & Gangjee, A. (2022). Design, Synthesis, and Biological Evaluation of 5,6,7,8-Tetrahydrobenzo[4,5]thieno[2,3-d]pyrimidines as Microtubule Targeting Agents. Molecules, 27(1), 321. https://doi.org/10.3390/molecules27010321