
Synthesis of PP2A-Activating PF-543 Derivatives and Investigation of Their Inhibitory Effects on Pancreatic Cancer Cells

 , and
, and
Abstract
:1. Introduction
2. Results and Discussion
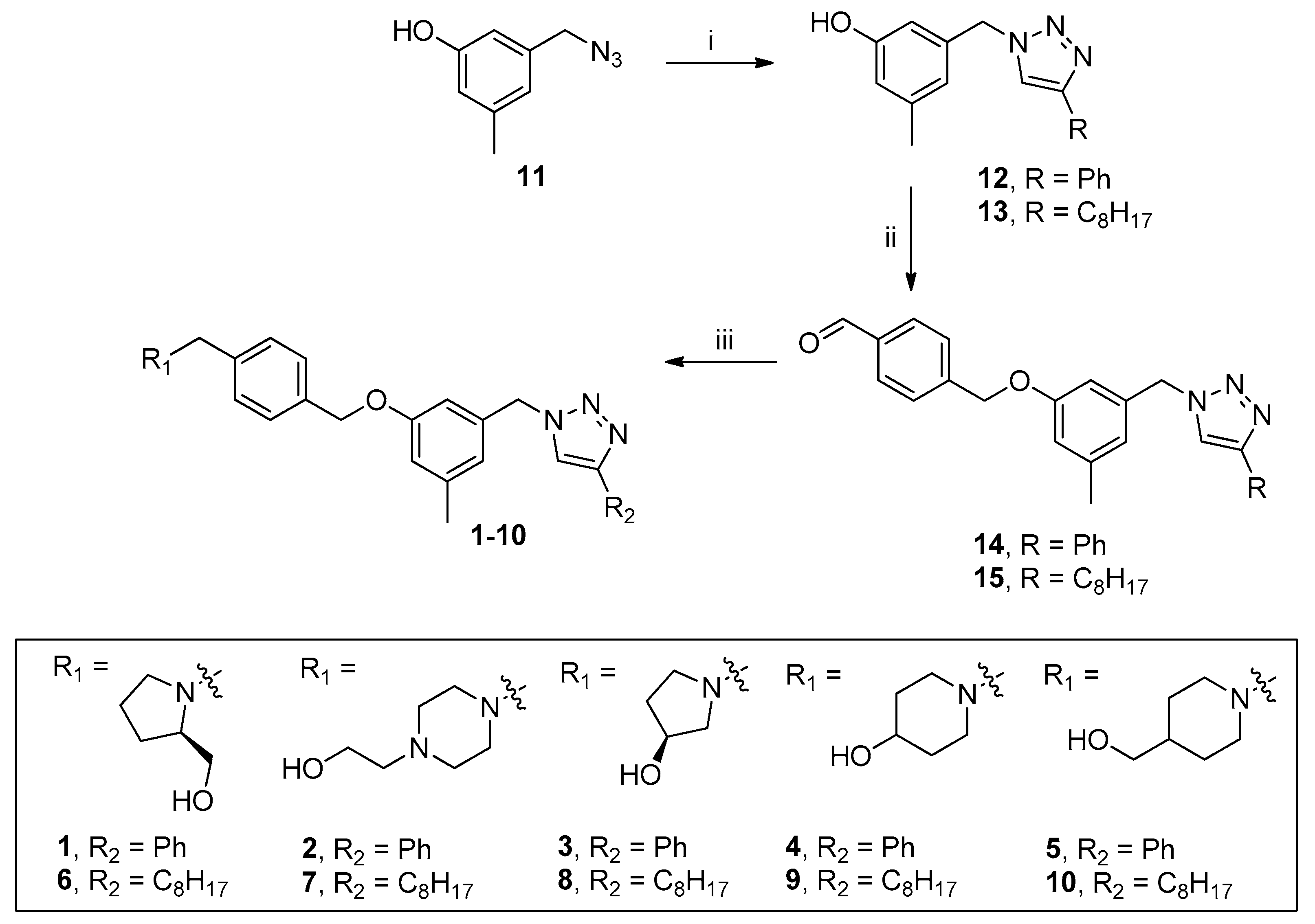
2.1. Chemical Synthesis
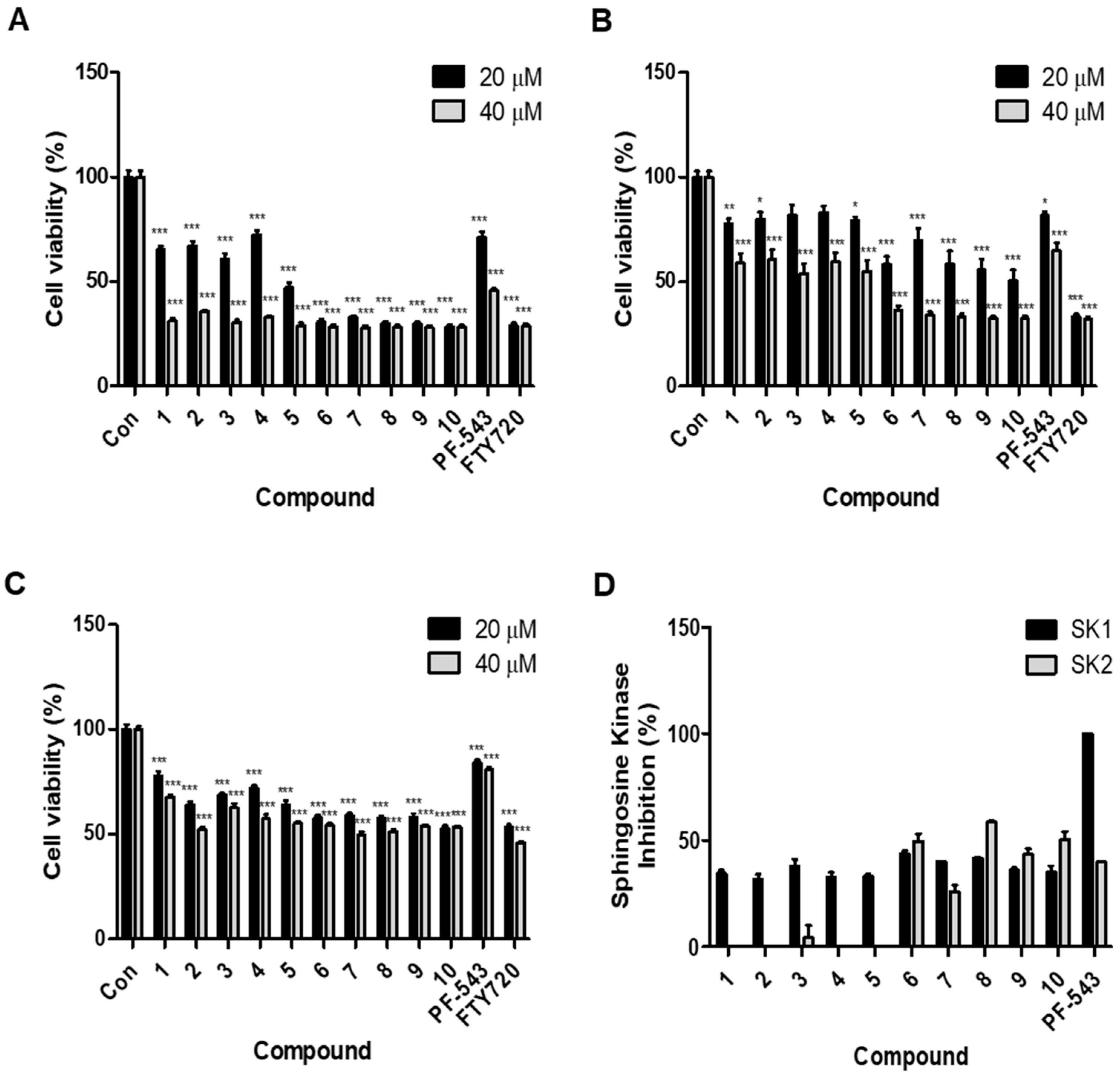
2.2. Compounds with an Aliphatic Tail Structure Showed a Stronger Cytotoxic Effect Than Compounds with an Aromatic Tail Structure
2.3. Compounds with an Aromatic Tail Structure Similar to PF-543 Have Selectivity for SK1 Inhibition
2.4. Compound 10 Effectively Reduces S1P and Sphingosine Levels
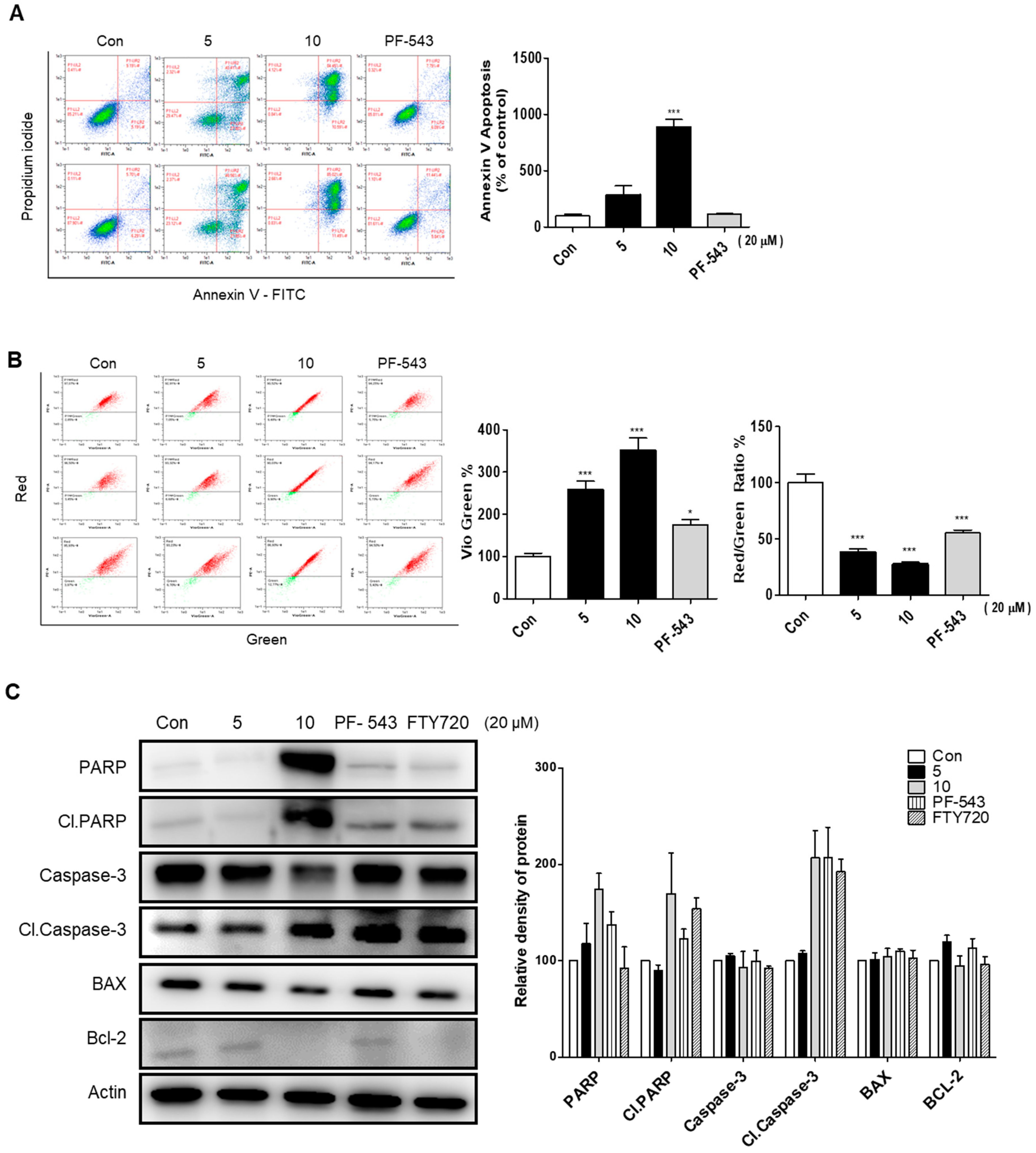
2.5. Compound 10 Shows a Stronger Apoptosis Effect Compared to Compound 5
2.6. Compound 10 with an Aliphatic Tail Activates PP2A
2.7. Compound 10 Shows Relatively Superior Metabolic Stability Compared to Compound 5
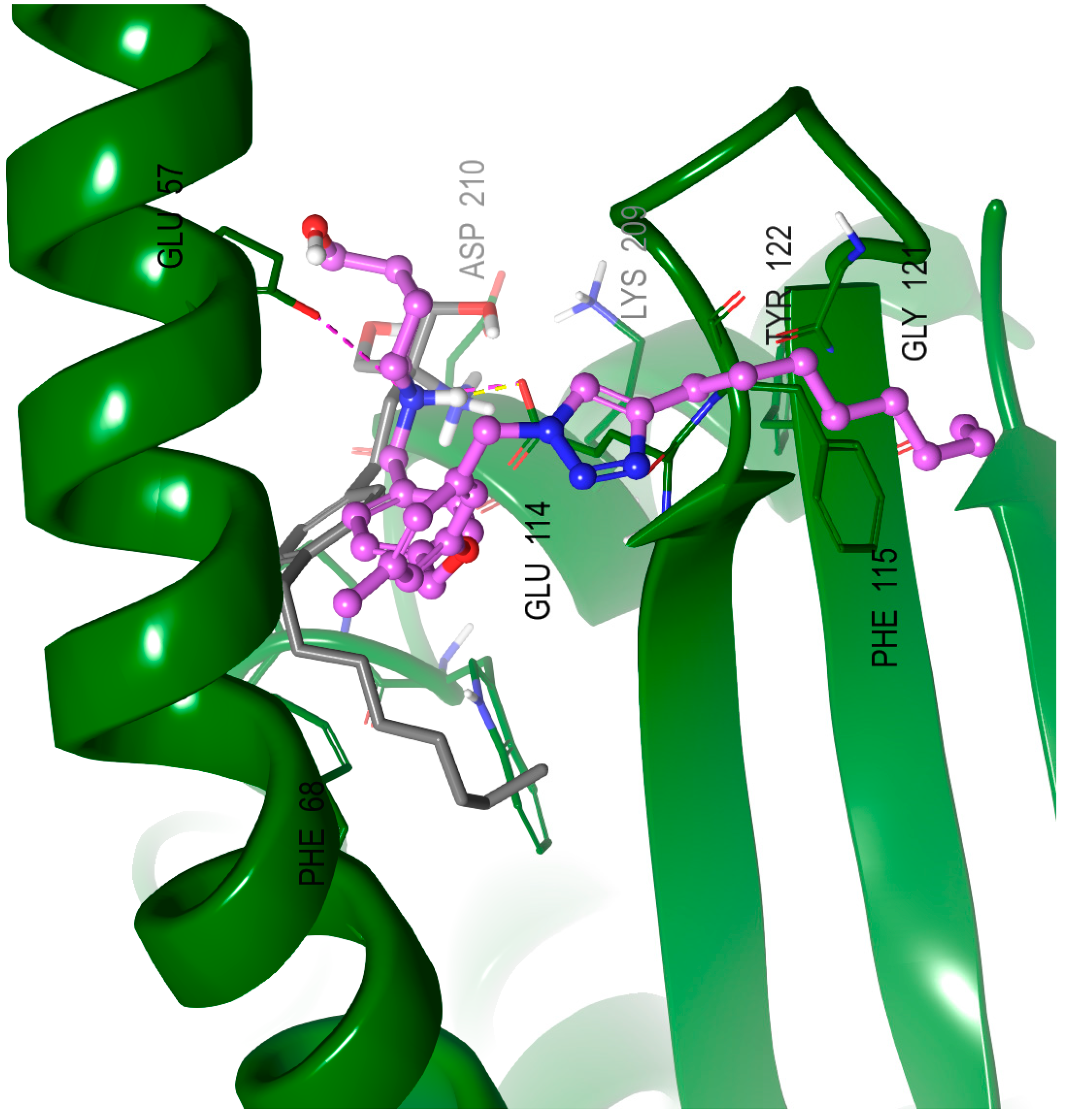
2.8. Molecular Modeling Studies
3. Experimental Section
3.1. Synthesis in General
3.2. Chemical Synthesis
3.3. Chemicals and Reagents
3.4. Sphingosine Kinase Activity Assay
3.5. Cell Culture and Proliferation Assays
3.6. Ceramide, Sphingosine, S1P Level
3.7. Annexin-V Staining of PF-543, Derivative 5, and 10
3.8. Mitochondrial Membrane Potential (MMP, Δψm)
3.9. PP2A Activity Assay
3.10. Western Blot Analysis
3.11. In Vitro Metabolic Stability of PF-543, Derivative 5, and 10
3.12. Molecular Modeling Studies
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Pitman, M.R.; Costabile, M.; Pitson, S.M. Recent advances in the development of sphingosine kinase inhibitors. Cell. Signal. 2016, 28, 1349–1363. [Google Scholar] [CrossRef] [PubMed]
- Grbčić, P.; Eichmann, T.O.; Kraljević Pavelić, S. The Sphingosine Kinase 2 Inhibitor ABC294640 Restores the Sensitivity of BRAFV600E Mutant Colon Cancer Cells to Vemurafenib by Reducing AKT-Mediated Expression of Nucleophosmin and Translationally-Controlled Tumour Protein. Int. J. Mol. Sci. 2021, 22, 10767. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cheng, L.; Shi, X.; Song, Y.; Chen, X.Y.; Chen, M.B.; Yao, J.; Zhang, Z.Q.; Cai, S. The sphingosine kinase inhibitor SKI-V suppresses cervical cancer cell growth. Int. J. Biol. Sci. 2022, 18, 2994–3005. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Mohammad, T.; Gupta, P.; Dahiya, R.; Parveen, S.; Luqman, S.; Hasan, G.M.; Hassan, M.I. Discovery of Harmaline as a Potent Inhibitor of Sphingosine Kinase-1: A Chemopreventive Role in Lung Cancer. ACS Omega 2020, 5, 21550–21560. [Google Scholar] [CrossRef] [PubMed]
- Hatoum, D.; Haddadi, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: Challenges for SphK as an oncotarget. Oncotarget 2017, 8, 36898–36929. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.M.; Yao, G.Q.; Siu, E. An Unanticipated Role for Sphingosine Kinase-2 in Bone and in the Anabolic Effect of Parathyroid Hormone. Endocrinology 2021, 162, bqab042. [Google Scholar] [CrossRef]
- LeBlanc, F.R.; Pearson, J.M.; Tan, S.F.; Cheon, H. Sphingosine kinase-2 is overexpressed in large granular lymphocyte leukaemia and promotes survival through Mcl-1. Br. J. Haematol. 2020, 190, 405–417. [Google Scholar] [CrossRef]
- Zheng, X.; Li, W.; Ren, L.; Liu, J.; Pang, X.; Chen, X.; Kang, D.; Wang, J.; Du, G. The sphingosine kinase-1/sphingosine-1-phosphate axis in cancer: Potential target for anticancer therapy. Pharmacol. Ther. 2019, 195, 85–99. [Google Scholar] [CrossRef]
- Hara-Yokoyama, M.; Terasawa, K.; Ichinose, S.; Watanabe, A.; Podyma-Inoue, K.A.; Akiyoshi, K.; Igarashi, Y.; Yanagishita, M. Sphingosine kinase 2 inhibitor SG-12 induces apoptosis via phosphorylation by sphingosine kinase 2. Bioorg. Med. Chem. Lett. 2013, 23, 2220–2224. [Google Scholar] [CrossRef]
- Cao, M.; Ji, C.; Zhou, Y.; Huang, W.; Ni, W.; Tong, X.; Wei, J.F. Sphingosine kinase inhibitors: A patent review. Int. J. Mol. Med. 2018, 41, 2450–2460. [Google Scholar] [CrossRef]
- Chun, J.; Brinkmann, V. A mechanistically novel, first oral therapy for multiple sclerosis: The development of fingolimod (FTY720, Gilenya). Discov. Med. 2011, 12, 213–228. [Google Scholar] [PubMed]
- Lim, K.G.; Sun, C.; Bittman, R.; Pyne, N.J.; Pyne, S. (R)-FTY720 methyl ether is a specific sphingosine kinase 2 inhibitor: Effect on sphingosine kinase 2 expression in HEK 293 cells and actin rearrangement and survival of MCF-7 breast cancer cells. Cell. Signal. 2011, 23, 1590–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Wang, J.; Zhao, Y.; Wu, Y.; Kwak, K.J.; Chen, C.-S.; Byrd, J.C.; Lee, R.J.; Phelps, M.A.; Lee, L.J.; et al. A novel liposomal formulation of FTY720 (Fingolimod) for promising enhanced targeted delivery. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 393–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Ju, T.; Gao, D.; Fang, Z.Y. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem. Biophys. Res. Commun. 2016, 470, 728–734. [Google Scholar] [CrossRef]
- Schnute, M.E.; McReynolds, M.D.; Carroll, J.; Chrencik, J.; Highkin, M.K.; Iyanar, K.; Jerome, G.; Rains, J.W.; Saabye, M.; Scholten, J.A.; et al. Discovery of a potent and selective sphingosine kinase 1 inhibitor through the molecular combination of chemotype-distinct screening hits. J. Med. Chem. 2017, 60, 2562–2572. [Google Scholar] [CrossRef]
- Kim, S.B.; Lee, T.; Moon, H.S.; Ki, S.H.; Oh, Y.S.; Lee, J.Y.; Kim, S.B.; Park, J.E.; Kwon, Y.; Kim, S.; et al. Verification of the necessity of the tolyl group of PF-543 for sphingosine kinase 1 inhibitory activity. Molecules 2020, 25, 2484. [Google Scholar] [CrossRef]
- Worrell, B.L.; Brown, A.M.; Santos, W.L.; Bevan, D.R. In silico characterization of structural distinctions between isoforms of human and mouse sphingosine kinases for accelerating drug discovery. J. Chem. Inf. Model. 2019, 59, 2339–2351. [Google Scholar] [CrossRef]
- Adams, D.R.; Tawati, S.; Berretta, G.; Rivas, P.L.; Baiget, J.; Jiang, Z.; Alsfouk, A.; Mackay, S.P.; Pyne, N.J.; Pyne, S. Topographical mapping of isoform-selectivity determinants for J-channel-binding inhibitors of sphingosine kinases 1 and 2. J. Med. Chem. 2019, 62, 3658–3676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.; Carmona, A.V.; Tiwari, A.K.; Trippier, P.C. Amide Bond Bioisosteres: Strategies, Synthesis, and Successes. J. Med. Chem. 2020, 63, 12290–12358. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Decker, A.M.; Laudermilk, L.; Maitra, R. Evaluation of Amide Bioisosteres Leading to 1,2,3-Triazole Containing Compounds as GPR88 Agonists: Design, Synthesis, and Structure-Activity Relationship Studies. J. Med. Chem. 2021, 64, 12397–12413. [Google Scholar] [CrossRef]
- Shrestha, J.; Hwang, G.T.; Lee, T.; Kim, S.W.; Oh, Y.S.; Kwon, Y.; Hong, S.W.; Kim, S.; Seop Moon, H.; Baek, D.J.; et al. Synthesis and biological evaluation of BODIPY-PF-543. Molecules 2019, 24, 4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Human (%) | Dog (%) | Rat (%) | Mouse (%) |
|---|---|---|---|---|
| 5 | 45.4 | 40.8 | 27.6 | 17.9 |
| 10 | 66.7 | 58.3 | 53.3 | 33.3 |
| PF-543 | 5.8 | 2.9 | 4.7 | 7.5 |
| Verapamil | 9.1 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.B.; Oh, Y.S.; Kim, K.J.; Cho, S.W.; Park, S.K.; Baek, D.J.; Park, E.-Y. Synthesis of PP2A-Activating PF-543 Derivatives and Investigation of Their Inhibitory Effects on Pancreatic Cancer Cells. Molecules 2022, 27, 3346. https://doi.org/10.3390/molecules27103346
Kim SB, Oh YS, Kim KJ, Cho SW, Park SK, Baek DJ, Park E-Y. Synthesis of PP2A-Activating PF-543 Derivatives and Investigation of Their Inhibitory Effects on Pancreatic Cancer Cells. Molecules. 2022; 27(10):3346. https://doi.org/10.3390/molecules27103346
Chicago/Turabian StyleKim, Su Bin, Yoon Sin Oh, Kwang Joon Kim, Sung Woo Cho, Seung Ki Park, Dong Jae Baek, and Eun-Young Park. 2022. "Synthesis of PP2A-Activating PF-543 Derivatives and Investigation of Their Inhibitory Effects on Pancreatic Cancer Cells" Molecules 27, no. 10: 3346. https://doi.org/10.3390/molecules27103346
APA StyleKim, S. B., Oh, Y. S., Kim, K. J., Cho, S. W., Park, S. K., Baek, D. J., & Park, E. -Y. (2022). Synthesis of PP2A-Activating PF-543 Derivatives and Investigation of Their Inhibitory Effects on Pancreatic Cancer Cells. Molecules, 27(10), 3346. https://doi.org/10.3390/molecules27103346