
Direct Utilization of Near-Infrared Light for Photooxidation with a Metal-Free Photocatalyst


Abstract
:
1. Introduction
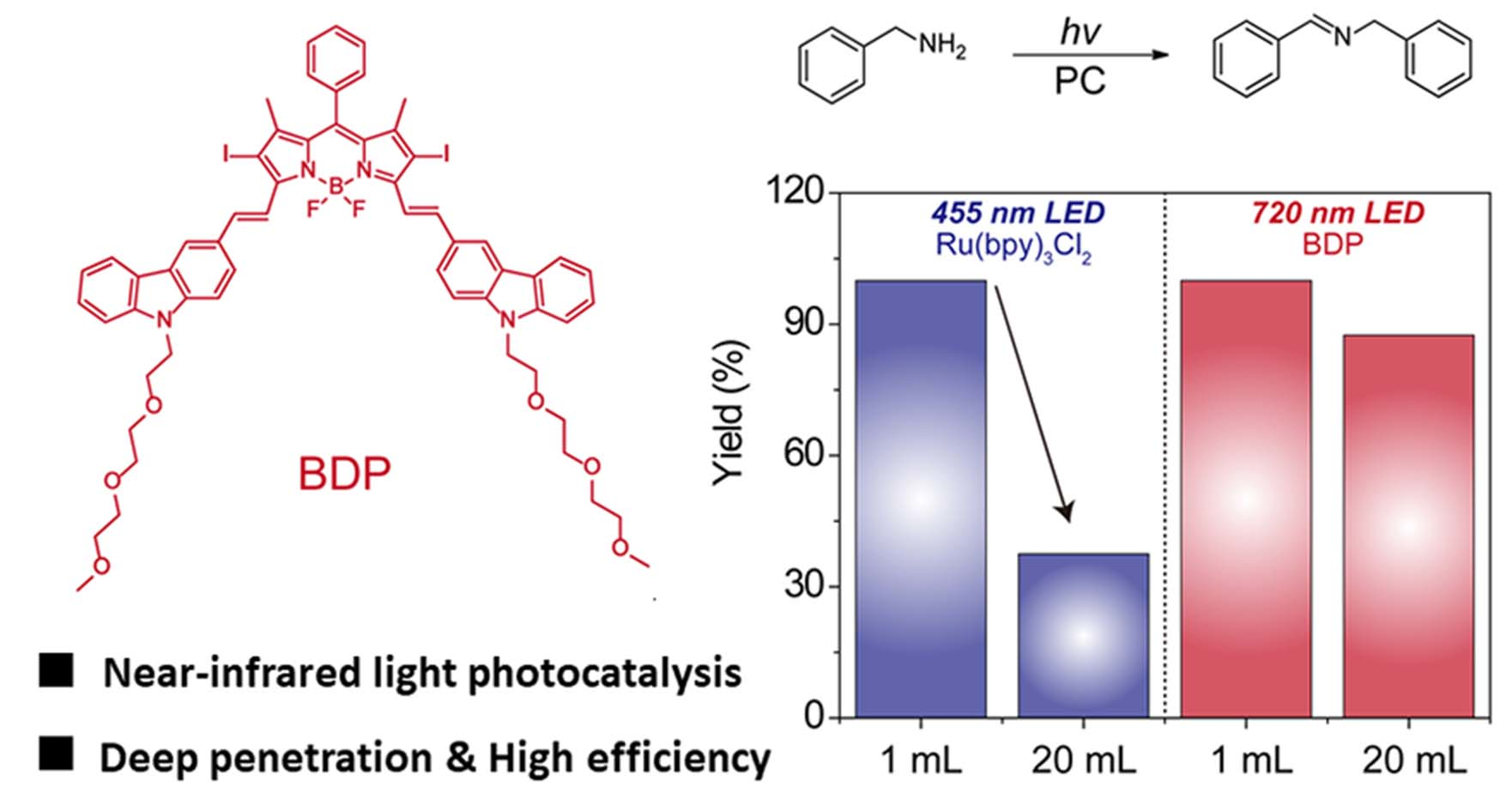
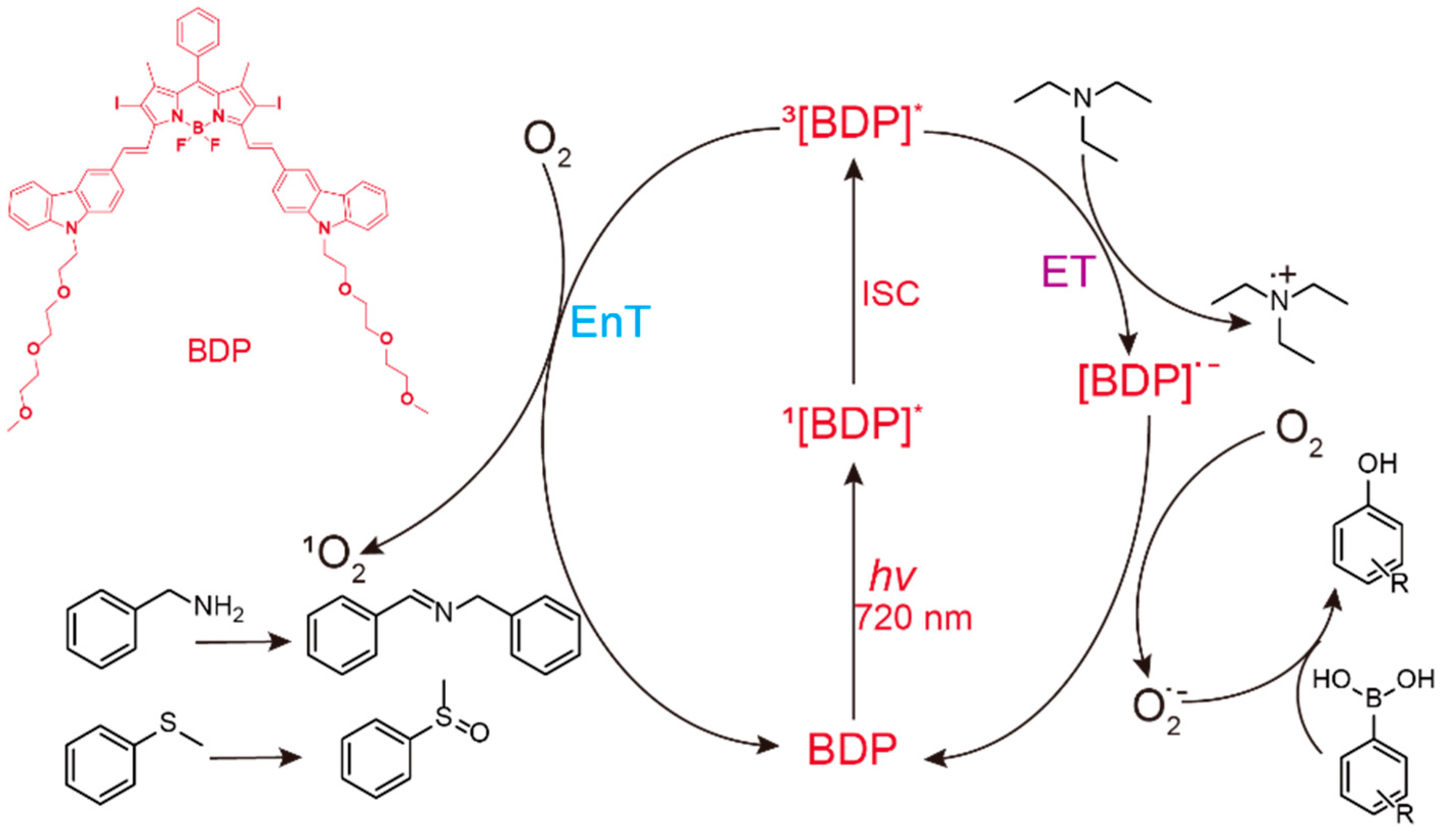
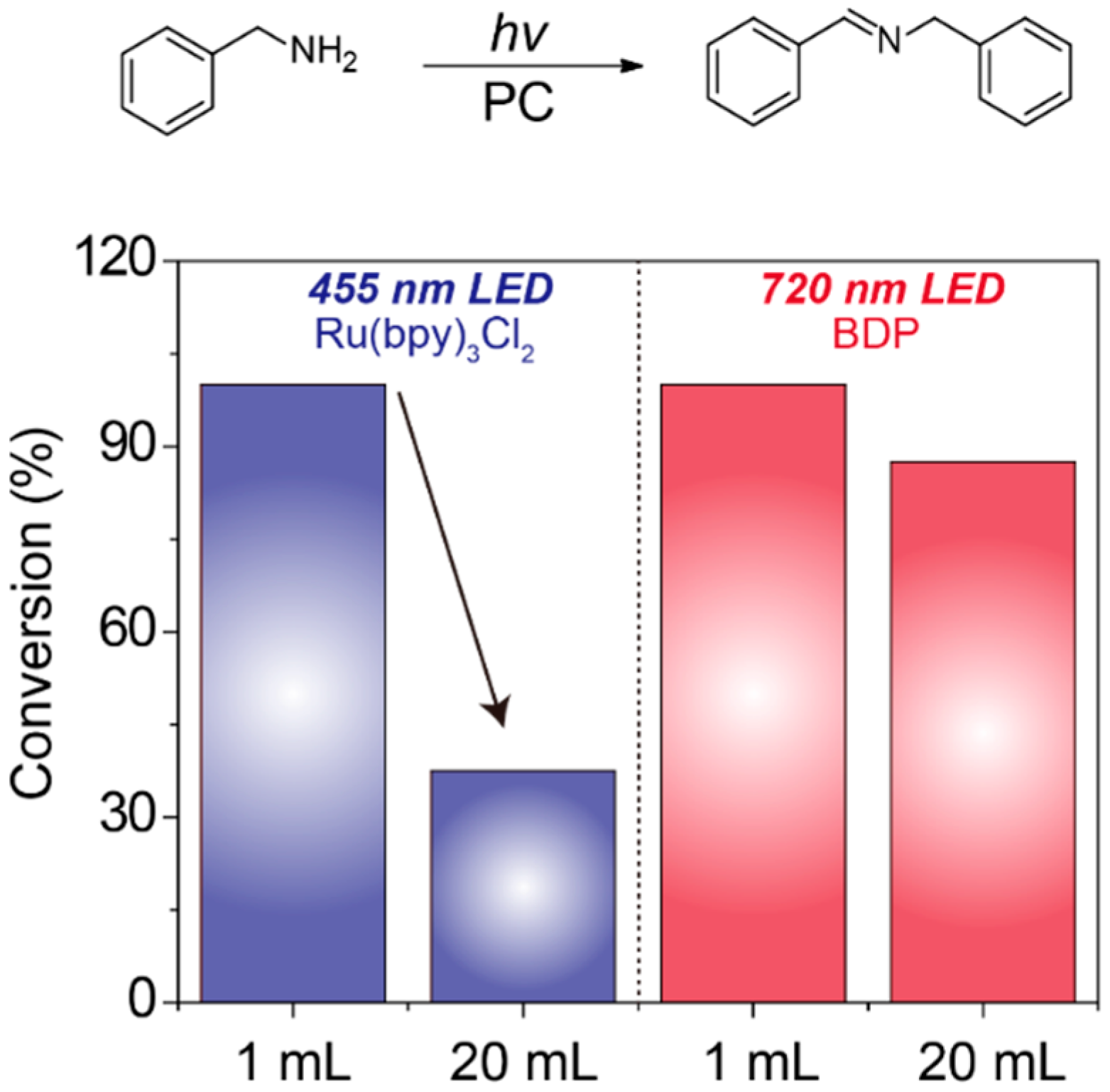
2. Results and Discussion
3. Materials and Methods
3.1. Measurement of Singlet Oxygen Generation Using DPBF as the Indicator
3.2. Photooxidation of Benzylamine Catalyzed by BDP with NIR Light Illumination
3.3. The Setup of the Large-Scale Reactions
3.4. Photooxidation of Sulfides Catalyzed by BDP with NIR-Light Illumination
3.5. Photooxidation of Phenylboronic Acids Catalyzed by BDP with NIR-Light Illumination
3.6. Turnover Number (TON) and Turnover Frequency (TOF) Calculation
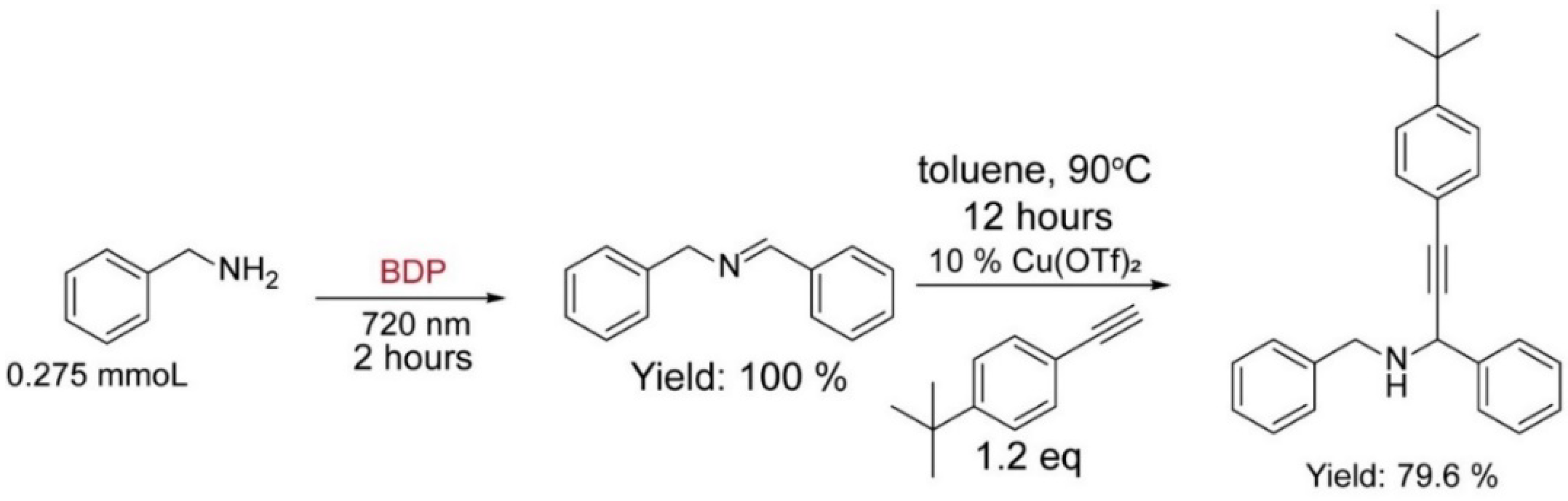
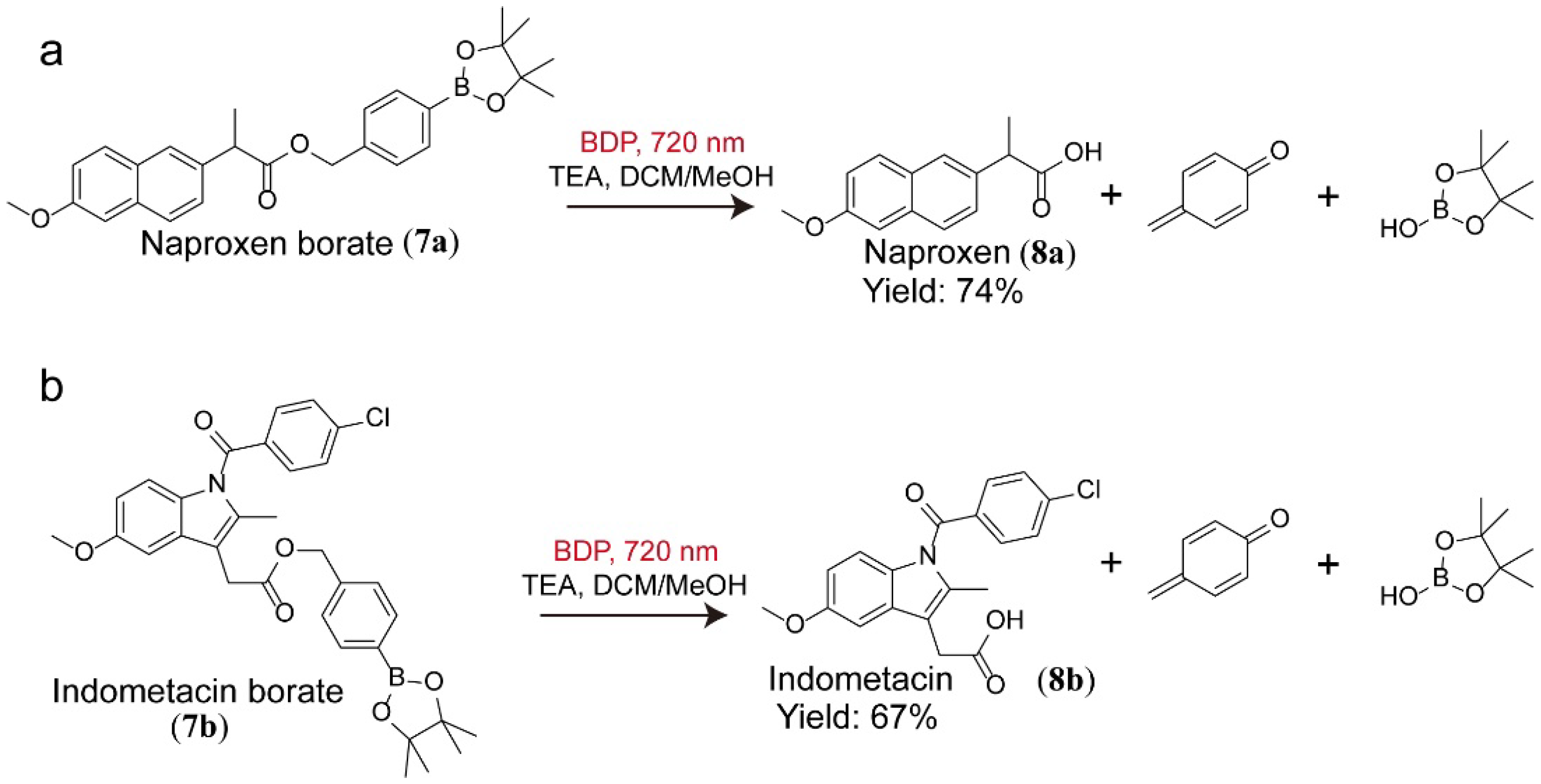
3.7. Tandem Reaction Catalyzed by BDP and Copper Salt
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ravelli, D.; Dondi, D.; Fagnoni, M.; Albini, A. Photocatalysis. A multi-faceted concept for green chemistry. Chem. Soc. Rev. 2009, 38, 1999. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, N.; Shanmugam, S.; Xu, J.; Boyer, C. Photocatalysis in organic and polymer synthesis. Chem. Soc. Rev. 2016, 45, 6165–6212. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szaciłowski, K.; Macyk, W.; Drzewiecka-Matuszek, A.; Brindell, M.; Stochel, G. Bioinorganic Photochemistry: Frontiers and Mechanisms. Chem. Rev. 2005, 105, 2647–2694. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Kanagaraj, K.; Fan, C.; Ji, J.; Xiao, C.; Wei, X.; Wu, W.; Yang, C. Photocatalytic Supramolecular Enantiodifferentiating Dimerization of 2-Anthracenecarboxylic Acid through Triplet–Triplet Annihilation. Org. Lett. 2018, 20, 1680–1683. [Google Scholar] [CrossRef]
- Rao, M.; Fan, C.; Ji, J.; Liang, W.; Wei, L.; Zhang, D.; Yan, Z.; Wu, W.; Yang, C. Catalytic Chiral Photochemistry Sensitized by Chiral Hosts-Grafted Upconverted Nanoparticles. ACS Appl. Mater. Interfaces 2022, 14, 21453–21460. [Google Scholar] [CrossRef] [PubMed]
- Ravetz, B.D.; Pun, A.B.; Churchill, E.M.; Congreve, D.N.; Rovis, T.; Campos, L.M. Photoredox catalysis using infrared light via triplet fusion upconversion. Nature 2019, 565, 343–346. [Google Scholar] [CrossRef]
- Ravetz, B.D.; Tay, N.E.S.; Joe, C.L.; Sezen-Edmonds, M.; Schmidt, M.A.; Tan, Y.; Janey, J.M.; Eastgate, M.D.; Rovis, T. Development of a Platform for Near-Infrared Photoredox Catalysis. ACS Cent. Sci. 2020, 6, 2053–2059. [Google Scholar] [CrossRef]
- Sellet, N.; Cormier, M.; Goddard, J.-P. The dark side of photocatalysis: Near-infrared photoredox catalysis for organic synthesis. Org. Chem. Front. 2021, 8, 6783–6790. [Google Scholar] [CrossRef]
- Singh-Rachford, T.N.; Castellano, F.N. Photon upconversion based on sensitized triplet–triplet annihilation. Coord. Chem. Rev. 2010, 254, 2560–2573. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, Q.; Feng, W.; Sun, Y.; Li, F. Upconversion Luminescent Materials: Advances and Applications. Chem. Rev. 2015, 115, 395–465. [Google Scholar] [CrossRef] [PubMed]
- Obah Kosso, A.R.; Sellet, N.; Baralle, A.; Cormier, M.; Goddard, J.-P. Cyanine-based near infra-red organic photoredox catalysis. Chem. Sci. 2021, 12, 6964–6968. [Google Scholar] [CrossRef] [PubMed]
- Loudet, A.; Burgess, K. BODIPY Dyes and Their Derivatives: Syntheses and Spectroscopic Properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, K.; Yang, W.; Wang, Z.; Zhong, F. The triplet excited state of Bodipy: Formation, modulation and application. Chem. Soc. Rev. 2015, 44, 8904–8939. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Li, Z.; Zhao, Y.; Zhang, Y.; Wu, S.; Zhao, J.; Han, G. Ultralow-Power Near Infrared Lamp Light Operable Targeted Organic Nanoparticle Photodynamic Therapy. J. Am. Chem. Soc. 2016, 138, 14586–14591. [Google Scholar] [CrossRef]
- Su, F.; Mathew, S.C.; Möhlmann, L.; Antonietti, M.; Wang, X.; Blechert, S. Aerobic Oxidative Coupling of Amines by Carbon Nitride Photocatalysis with Visible Light. Angew. Chem. Int. Ed. 2011, 50, 657–660. [Google Scholar] [CrossRef]
- Lang, X.; Ji, H.; Chen, C.; Ma, W.; Zhao, J. Selective Formation of Imines by Aerobic Photocatalytic Oxidation of Amines on TiO2. Angew. Chem. Int. Ed. 2011, 50, 3934–3937. [Google Scholar] [CrossRef]
- Rueping, M.; Vila, C.; Szadkowska, A.; Koenigs, R.M.; Fronert, J. Photoredox Catalysis as an Efficient Tool for the Aerobic Oxidation of Amines and Alcohols: Bioinspired Demethylations and Condensations. ACS Catal. 2012, 2, 2810–2815. [Google Scholar] [CrossRef]
- Beatty, J.W.; Stephenson, C.R.J. Amine Functionalization via Oxidative Photoredox Catalysis: Methodology Development and Complex Molecule Synthesis. Acc. Chem. Res. 2015, 48, 1474–1484. [Google Scholar] [CrossRef] [Green Version]
- Cozzi, P.G. Metal–Salen Schiff base complexes in catalysis: Practical aspects. Chem. Soc. Rev. 2004, 33, 410–421. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Li, J. Molecular Assembly of Schiff Base Interactions: Construction and Application. Chem. Rev. 2015, 115, 1597–1621. [Google Scholar] [CrossRef] [PubMed]
- Segura, J.L.; Mancheño, M.J.; Zamora, F. Covalent organic frameworks based on Schiff-base chemistry: Synthesis, properties and potential applications. Chem. Soc. Rev. 2016, 45, 5635–5671. [Google Scholar] [CrossRef] [PubMed]
- Farney, E.P.; Chapman, S.J.; Swords, W.B.; Torelli, M.D.; Hamers, R.J.; Yoon, T.P. Discovery and Elucidation of Counteranion Dependence in Photoredox Catalysis. J. Am. Chem. Soc. 2019, 141, 6385–6391. [Google Scholar] [CrossRef]
- Nosaka, Y.; Nosaka, A.Y. Generation and Detection of Reactive Oxygen Species in Photocatalysis. Chem. Rev. 2017, 117, 11302–11336. [Google Scholar] [CrossRef] [PubMed]
- Merkel, P.B.; Nilsson, R.; Kearns, D.R. Deuterium effects on singlet oxygen lifetimes in solutions. New test of singlet oxygen reactions. J. Am. Chem. Soc. 1972, 94, 1030–1031. [Google Scholar] [CrossRef]
- Meyet, C.E.; Pierce, C.J.; Larsen, C.H. A Single Cu(II) Catalyst for the Three-Component Coupling of Diverse Nitrogen Sources with Aldehydes and Alkynes. Org. Lett. 2012, 14, 964–967. [Google Scholar] [CrossRef]
- Lang, X.; Zhao, J.; Chen, X. Cooperative photoredox catalysis. Chem. Soc. Rev. 2016, 45, 3026–3038. [Google Scholar] [CrossRef]
- Carreno, M.C. Applications of Sulfoxides to Asymmetric Synthesis of Biologically Active Compounds. Chem. Rev. 1995, 95, 1717–1760. [Google Scholar] [CrossRef]
- Evans, D.A.; Andrews, G.C. Allylic sulfoxides. Useful intermediates in organic synthesis. Acc. Chem. Res. 1974, 7, 147–155. [Google Scholar] [CrossRef]
- Fernández, I.; Khiar, N. Recent Developments in the Synthesis and Utilization of Chiral Sulfoxides. Chem. Rev. 2003, 103, 3651–3706. [Google Scholar] [CrossRef] [PubMed]
- Sipos, G.; Drinkel, E.E.; Dorta, R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.J.; Gu, C.L.; Kacher, M.L.; Foote, C.S. Chemistry of singlet oxygen. 45. Mechanism of the photooxidation of sulfides. J. Am. Chem. Soc. 1983, 105, 4717–4721. [Google Scholar] [CrossRef]
- Zou, Y.-Q.; Chen, J.-R.; Liu, X.-P.; Lu, L.-Q.; Davis, R.L.; Jørgensen, K.A.; Xiao, W.-J. Highly Efficient Aerobic Oxidative Hydroxylation of Arylboronic Acids: Photoredox Catalysis Using Visible Light. Angew. Chem. Int. Ed. 2012, 51, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Pitre, S.P.; McTiernan, C.D.; Ismaili, H.; Scaiano, J.C. Mechanistic Insights and Kinetic Analysis for the Oxidative Hydroxylation of Arylboronic Acids by Visible Light Photoredox Catalysis: A Metal-Free Alternative. J. Am. Chem. Soc. 2013, 135, 13286–13289. [Google Scholar] [CrossRef]
- Klán, P.; Šolomek, T.; Bochet, C.G.; Blanc, A.; Givens, R.; Rubina, M.; Popik, V.; Kostikov, A.; Wirz, J. Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy. Chem. Rev. 2013, 113, 119–191. [Google Scholar] [CrossRef]
- Shao, Q.; Xing, B. Photoactive molecules for applications in molecular imaging and cell biology. Chem. Soc. Rev. 2010, 39, 2835–2846. [Google Scholar] [CrossRef]
- Huang, L.; Zeng, L.; Chen, Y.; Yu, N.; Wang, L.; Huang, K.; Zhao, Y.; Han, G. Long wavelength single photon like driven photolysis via triplet triplet annihilation. Nat. Commun. 2021, 12, 122. [Google Scholar] [CrossRef]
- Qiu, F.-Y.; Zhang, M.; Ji, R.; Du, F.-S.; Li, Z.-C. Oxidation-Responsive Polymer–Drug Conjugates with a Phenylboronic Ester Linker. Macromol. Rapid Commun. 2015, 36, 2012–2018. [Google Scholar] [CrossRef]
- Peiró Cadahía, J.; Previtali, V.; Troelsen, N.S.; Clausen, M.H. Prodrug strategies for targeted therapy triggered by reactive oxygen species. Med. Chem. Commun. 2019, 10, 1531–1549. [Google Scholar] [CrossRef]
- Mitsunaga, M.; Ogawa, M.; Kosaka, N.; Rosenblum, L.T.; Choyke, P.L.; Kobayashi, H. Cancer cell–selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med. 2011, 17, 1685–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeForest, C.A.; Anseth, K.S. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem. 2011, 3, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, L.G.; Pedersen, C.M.; Bols, M. Safe radical azidonation using polystyrene supported diazidoiodate(I). Tetrahedron 2005, 61, 123–127. [Google Scholar] [CrossRef]
- Raza, F.; Park, J.H.; Lee, H.-R.; Kim, H.-I.; Jeon, S.-J.; Kim, J.-H. Visible-Light-Driven Oxidative Coupling Reactions of Amines by Photoactive WS2 Nanosheets. ACS Catal. 2016, 6, 2754–2759. [Google Scholar] [CrossRef]
- Patil, R.D.; Adimurthy, S. Copper-Catalyzed Aerobic Oxidation of Amines to Imines under Neat Conditions with Low Catalyst Loading. Adv. Synth. Catal. 2011, 353, 1695–1700. [Google Scholar] [CrossRef]
- Su, C.; Tandiana, R.; Tian, B.; Sengupta, A.; Tang, W.; Su, J.; Loh, K.P. Visible-Light Photocatalysis of Aerobic Oxidation Reactions Using Carbazolic Conjugated Microporous Polymers. ACS Catal. 2016, 6, 3594–3599. [Google Scholar] [CrossRef]
- Bradamante, S.; Colombo, S.; Pagani, G.A.; Roelens, S. The Reaction of Pyruvic Acid with Amines and Aminoesters Reexamined. Preliminary communication. Helv. Chim. Acta 1981, 64, 568–571. [Google Scholar] [CrossRef]
- Sasamoto, N.; Dubs, C.; Hamashima, Y.; Sodeoka, M. Pd(II)-Catalyzed Asymmetric Addition of Malonates to Dihydroisoquinolines. J. Am. Chem. Soc. 2006, 128, 14010–14011. [Google Scholar] [CrossRef]
- Wu, X.; Gorden, A.E.V. 2-Quinoxalinol Salen Copper Complexes for Oxidation of Aryl Methylenes. Eur. J. Org. Chem. 2009, 2009, 503–509. [Google Scholar] [CrossRef]
- Sharma, A.; Sharma, N.; Kumar, R.; Sharma, U.K.; Sinha, A.K. Water-promoted cascade synthesis of α-arylaldehydes from arylalkenes using N-halosuccinimides: An avenue for asymmetric oxidation using Cinchona organocatalysis. Chem. Commun. 2009, 5299–5301. [Google Scholar] [CrossRef]
- Kasaya, Y.; Hoshi, K.; Terada, Y.; Nishida, A.; Shuto, S.; Arisawa, M. Aromatic Enamide/Ene Metathesis toward Substituted Indoles and Its Application to the Synthesis of Indomethacins. Eur. J. Org. Chem. 2009, 2009, 4606–4613. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | Conversion b | TON c | TOF (min−1) d |
|---|---|---|---|---|---|
| 1 |  1a 1a |  2a 2a | 100 | 8.59 × 102 | 7.16 |
| 2 |  1b 1b |  2b 2b | 100 | 8.59 × 102 | 7.16 |
| 3 |  1c 1c |  2c 2c | 95.6 | 8.22 × 102 | 6.85 |
| 4 |  1d 1d |  2d 2d | 95.2 | 8.18 × 102 | 6.82 |
| 5 |  1e 1e |  2e 2e | 100 | 8.59 × 102 | 7.16 |
| 6 |  1f 1f |  2f 2f | 100 | 8.59 × 102 | 7.16 |
| 7 |  1g 1g |  2g 2g | 100 | 8.59 × 102 | 7.16 |
| 8 e |  1h 1h |  2h 2h | 76.9 | 6.61 × 102 | 5.51 |
| 9 |  1i 1i |  2i 2i | 58.8 | 5.05 × 102 | 4.21 |
| 10 |  1j 1j |  2j 2j | 100 | 8.59 × 102 | 7.16 |
| Entry | Substrate | Product | Conversion b | TON c | TOF (min−1) d |
|---|---|---|---|---|---|
| 1 |  3a 3a |  4a 4a | 100 | 430 | 3.6 |
| 2 |  3b 3b |  4b 4b | 100 | 430 | 3.6 |
| 3 |  3c 3c |  4c 4c | 96 | 413 | 3.5 |
| 4 |  3d 3d |  4d 4d | 90 | 387 | 3.2 |
| Entry | Substrate | Product | Yield b | TON c | TOF (min−1) d |
|---|---|---|---|---|---|
| 1 |  5a 5a |  6a 6a | 85 | 365 | 3.1 |
| 2 |  5b 5b |  6b 6b | 95 | 408 | 3.4 |
| 3 |  5c 5c |  6c 6c | 90 | 387 | 3.2 |
| 4 |  5d 5d |  6d 6d | 87 | 374 | 3.1 |
| 5 |  5e 5e |  6e 6e | 94 | 404 | 3.4 |
| 6 |  5f 5f |  6f 6f | 95 | 408 | 3.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, L.; Wang, Z.; Zhang, T.; Duan, C. Direct Utilization of Near-Infrared Light for Photooxidation with a Metal-Free Photocatalyst. Molecules 2022, 27, 4047. https://doi.org/10.3390/molecules27134047
Zeng L, Wang Z, Zhang T, Duan C. Direct Utilization of Near-Infrared Light for Photooxidation with a Metal-Free Photocatalyst. Molecules. 2022; 27(13):4047. https://doi.org/10.3390/molecules27134047
Chicago/Turabian StyleZeng, Le, Zhonghe Wang, Tiexin Zhang, and Chunying Duan. 2022. "Direct Utilization of Near-Infrared Light for Photooxidation with a Metal-Free Photocatalyst" Molecules 27, no. 13: 4047. https://doi.org/10.3390/molecules27134047
APA StyleZeng, L., Wang, Z., Zhang, T., & Duan, C. (2022). Direct Utilization of Near-Infrared Light for Photooxidation with a Metal-Free Photocatalyst. Molecules, 27(13), 4047. https://doi.org/10.3390/molecules27134047