
Insights into the Allosteric Effect of SENP1 Q597A Mutation on the Hydrolytic Reaction of SUMO1 via an Integrated Computational Study

Abstract
:1. Introduction
2. Results and Discussion
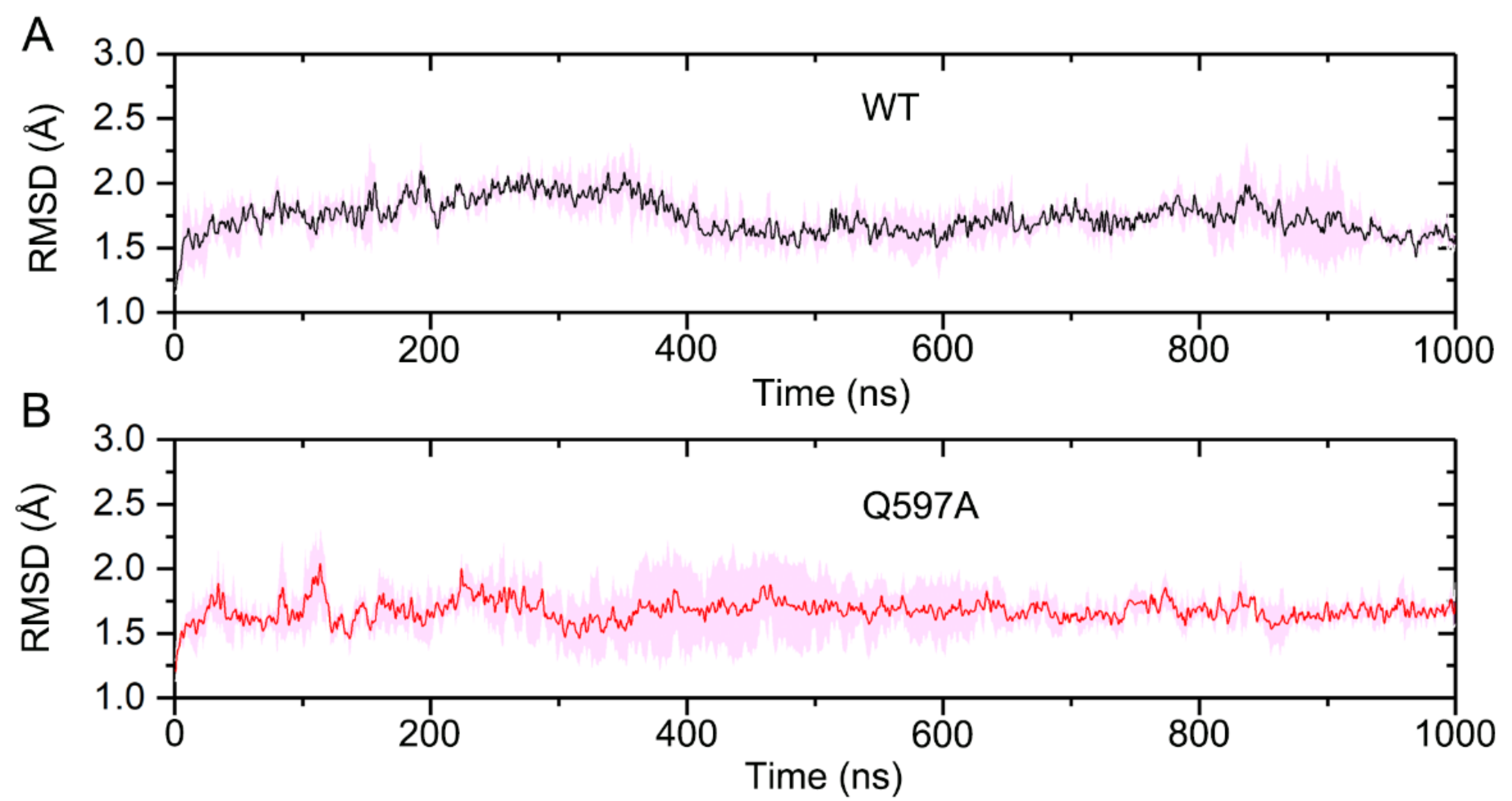
2.1. System Stability
2.2. Q597A Mutation Enhanced the Coupled Motions of Protein Domains
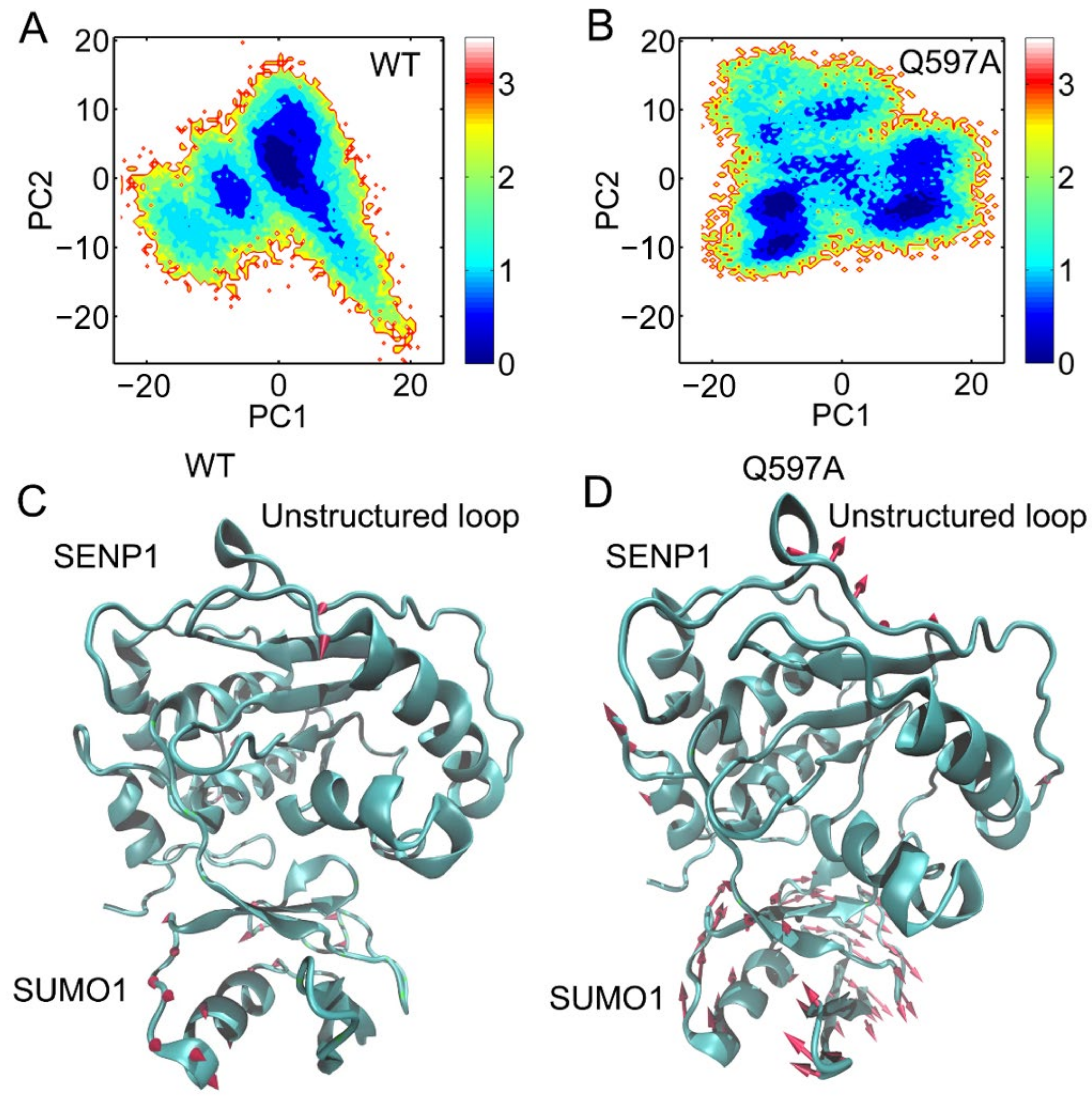
2.3. Pricipal Component Analysis (PCA)
2.4. Community Network Analysis
2.5. Q597A Mutation Caused Conformational Changes to Cys603 and His533
2.6. Q597A Mutation Had a Minor Effect on the Binding Affinity of SUMO1
3. Conclusions
4. Materials and Methods
4.1. Structural Preparation
4.2. MD Simulations
4.3. Principal Component Analysis (PCA)
4.4. Dynamic Cross-Correlation Analysis
4.5. MM–PBSA Binding Free Energy Calculations
4.6. Community Network Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Mukhopadhyay, D.; Dasso, M. Modification in reverse: The SUMO proteases. Trends Biochem. Sci. 2007, 32, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sharma, P.; Athanasiou, M.; Kumar, A.; Yamada, S.; Kuehn, M.R. Mutation of SENP1/SuPr-2 Reveals an Essential Role for Desumoylation in Mouse Development. Mol. Cell. Biol. 2005, 25, 5171–5182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Zhang, K.Y.J. Advances in the development of SUMO specific protease (SENP) inhibitors. Comput. Struct. Biotechnol. J. 2015, 13, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T.H. SUMO-Specific Protease 1 Is Critical for Early Lymphoid Development through Regulation of STAT5 Activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef]
- Cheng, J.; Bawa, T.; Lee, P.; Gong, L.; Yeh, E.T.H. Role of desumoylation in the development of prostate cancer. Neoplasia 2006, 8, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Cheng, J.K. Small ubiquitin-like modifier protein-specific protease 1 and prostate cancer. Asian J. Androl. 2009, 11, 36–38. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, S.; Jääskeläinen, T.; Karvonen, U.; Rytinki, M.M.; Makkonen, H.; Gioeli, D.; Paschal, B.M.; Palvimo, J.J. SUMO-specific protease 1 (SENP1) reverses the hormone-augmented SUMOylation of androgen receptor and modulates gene responses in prostate cancer cells. Mol. Endocrinol. 2009, 23, 292–307. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Xia, N.; Li, T.; Xu, Y.; Zou, Y.; Zuo, Y.; Fan, Q.; Bawa-Khalfe, T.; Yeh, E.T.H.; Cheng, J. SUMO-specific protease 1 promotes prostate cancer progression and metastasis. Oncogene 2013, 32, 2493–2498. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.P.; Wong, C.C.L.; Kai, A.K.L.; Ho, D.W.H.; Lau, E.Y.T.; Tsui, Y.M.; Chan, L.K.; Cheung, T.T.; Chok, K.S.H.; Chan, A.C.Y.; et al. SENP1 promotes hypoxia-induced cancer stemness by HIF-1α deSUMOylation and SENP1/HIF-1α positive feedback loop. Gut 2017, 66, 2149–2159. [Google Scholar] [CrossRef]
- Tao, Y.; Li, R.; Shen, C.; Li, J.; Zhang, Q.; Ma, Z.; Wang, F.; Wang, Z. SENP1 is a crucial promotor for hepatocellular carcinoma through deSUMOylation of UBE2T. Aging 2020, 12, 1563–1576. [Google Scholar] [CrossRef]
- Xu, Z.; Chau, S.F.; Lam, K.H.; Chan, H.Y.; Ng, T.B.; Au, S.W.N. Crystal structure of the SENP1 mutant C603S-SUMO complex reveals the hydrolytic mechanism of SUMO-specific protease. Biochem. J. 2006, 398, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.N.; Dong, C.; Liu, H.; Naismith, J.H.; Hay, R.T. The structure of SENP1-SUMO-2 complex suggests a structural basis for discrimination between SUMO paralogues during processing. Biochem. J. 2006, 397, 279–288. [Google Scholar] [CrossRef]
- Lu, S.; Shen, Q.; Zhang, J.; Shen, Q.; Zhang, J. Allosteric Methods and Their Applications: Facilitating the Discovery of Allosteric Drugs and the Investigation of Allosteric Mechanisms. Acc. Chem. Res. 2019, 52, 492–500. [Google Scholar] [CrossRef]
- Lu, S.; Ji, M.; Ni, D.; Zhang, J. Discovery of hidden allosteric sites as novel targets for allosteric drug design. Drug Discov. Today 2018, 23, 359–365. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Ni, D.; Zhang, J. Allosteric Modulator Discovery: From Serendipity to Structure-Based Design. J. Med. Chem. 2019, 62, 6405–6421. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; Wang, D.; Chen, Y.; Zhang, N. Recent Advances in the Discovery of CK2 Allosteric Inhibitors: From Traditional Screening to Structure-Based Design. Molecules 2020, 25, 870. [Google Scholar] [CrossRef] [Green Version]
- Maloney, R.C.; Zhang, M.; Jang, H.; Nussinov, R. The mechanism of activation of monomeric B-Raf V600E. Comput. Struct. Biotechnol. J. 2021, 19, 3349–3363. [Google Scholar] [CrossRef]
- Foutch, D.; Pham, B.; Shen, T. Protein conformational switch discerned via network centrality properties. Comput. Struct. Biotechnol. J. 2021, 19, 3599–3608. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.-J. Allostery in disease and in drug discovery. Cell 2013, 153, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Okeke, C.J.; Musyoka, T.M.; Sheik Amamuddy, O.; Barozi, V.; Tastan Bishop, Ö. Allosteric pockets and dynamic residue network hubs of falcipain 2 in mutations including those linked to artemisinin resistance. Comput. Struct. Biotechnol. J. 2021, 19, 5647–5666. [Google Scholar] [CrossRef]
- Hernández-Alvarez, L.; Oliveira, A.B.; Hernández-González, J.E.; Chahine, J.; Pascutti, P.G.; de Araujo, A.S.; de Souza, F.P. Computational study on the allosteric mechanism of Leishmania major IF4E-1 by 4E-interacting protein-1: Unravelling the determinants of m7GTP cap recognition. Comput. Struct. Biotechnol. J. 2021, 19, 2027–2044. [Google Scholar] [CrossRef]
- Zhang, M.; Jang, H.; Nussinov, R. The mechanism of PI3Kα activation at the atomic level. Chem. Sci. 2019, 10, 3671–3680. [Google Scholar] [CrossRef] [Green Version]
- An, X.; Bai, Q.; Bing, Z.; Liu, H.; Yao, X. Insights into the molecular mechanism of positive cooperativity between partial agonist MK-8666 and full allosteric agonist AP8 of hGPR40 by Gaussian accelerated molecular dynamics (GaMD) simulations. Comput. Struct. Biotechnol. J. 2021, 19, 3978–3989. [Google Scholar] [CrossRef]
- Han, D.; Wang, H.; Wujieti, B.; Zhang, B.; Cui, W.; Chen, B.Z. Insight into the drug resistance mechanisms of GS-9669 caused by mutations of HCV NS5B polymerase via molecular simulation. Comput. Struct. Biotechnol. J. 2021, 19, 2761–2774. [Google Scholar] [CrossRef]
- Hu, X.; Pang, J.; Zhang, J.; Shen, C.; Chai, X.; Wang, E.; Chen, H.; Wang, X.; Duan, M.; Fu, W.; et al. Discovery of Novel GR Ligands toward Druggable GR Antagonist Conformations Identified by MD Simulations and Markov State Model Analysis. Adv. Sci. 2022, 9, 2102435. [Google Scholar] [CrossRef]
- Lu, S.; He, X.; Yang, Z.; Chai, Z.; Zhou, S.; Wang, J.; Rehman, A.U.; Ni, D.; Pu, J.; Sun, J.; et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat. Commun. 2021, 12, 4721. [Google Scholar] [CrossRef]
- Lu, S.; Zhang, J. Small Molecule Allosteric Modulators of G-Protein-Coupled Receptors: Drug–Target Interactions. J. Med. Chem. 2019, 62, 24–45. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Liang, W.; Shi, X.; Fan, J.; Kong, R.; Liu, Y.; Zhang, J.; Chen, T.; Lu, S. Delineating the activation mechanism and conformational landscape of a class B G protein-coupled receptor glucagon receptor. Comput. Struct. Biotechnol. J. 2022, 20, 628–639. [Google Scholar] [CrossRef]
- Lu, S.; Ni, D.; Wang, C.; He, X.; Lin, H.; Wang, Z.; Zhang, J. Deactivation Pathway of Ras GTPase Underlies Conformational Substates as Targets for Drug Design. ACS Catal. 2019, 9, 7188–7196. [Google Scholar] [CrossRef]
- Li, X.; Dai, J.; Ni, D.; He, X.; Zhang, H.; Zhang, J.; Fu, Q.; Liu, Y.; Lu, S. Insight into the mechanism of allosteric activation of PI3Kα by oncoprotein K-Ras4B. Int. J. Biol. Macromol. 2020, 144, 643–655. [Google Scholar] [CrossRef]
- Wang, Y.; Ji, D.; Lei, C.; Chen, Y.; Qiu, Y.; Li, X.; Li, M.; Ni, D.; Pu, J.; Zhang, J.; et al. Mechanistic insights into the effect of phosphorylation on Ras conformational dynamics and its interactions with cell signaling proteins. Comput. Struct. Biotechnol. J. 2021, 19, 1184–1199. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Du, K.; Yuanhao, W.; Li, M.; Fan, J.; Ni, D.; Lu, S.; Biao, X.; Liu, Y. Autopromotion of K-Ras4B feedback activation through an SOS-mediated long-range allosteric effect. Front. Mol. Biosci. 2022, 9, 860962. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.; Peng, T.; Chai, Z.; Ni, D.; Liu, Y.; Zhang, J.; Chen, T.; Lu, S. Atomic-scale insights into allosteric inhibition and evolutional rescue mechanism of Streptococcus thermophilus Cas9 by the anti-CRISPR protein AcrIIA6. Comput. Struct. Biotechnol. J. 2021, 19, 6108–6124. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Fan, X.; Ji, D.; Wang, Y.; Fan, J.; Li, M.; Ni, D.; Lu, S.; Li, X.; Chai, Z. Elucidation of the conformational dynamics and assembly of Argonaute−RNA complexes by distinct yet coordinated actions of the supplementary microRNA. Comput. Struct. Biotechnol. J. 2022, 20, 1352–1365. [Google Scholar] [CrossRef]
- Ni, D.; Wei, J.; He, X.; Rehman, A.U.; Li, X.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem. Sci. 2021, 12, 464–476. [Google Scholar] [CrossRef]
- Lu, S.; Chen, Y.; Wei, J.; Zhao, M.; Ni, D.; He, X.; Zhang, J. Mechanism of allosteric activation of SIRT6 revealed by the action of rationally designed activators. Acta Pharm. Sin. B 2021, 11, 1355–1361. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, Y.; Ni, D.; Huang, Z.; Wei, J.; Feng, L.; Su, J.C.; Wei, Y.; Ning, S.; Yang, X.; et al. Targeting a cryptic allosteric site of SIRT6 with small-molecule inhibitors that inhibit the migration of pancreatic cancer cells. Acta Pharm. Sin. B 2022, 12, 876–889. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, M.; Li, M.; Ni, D.; Wang, Y.; Deng, L.; Du, K.; Lu, S.; Shi, H.; Cai, C. Mechanistic Insights Into Co-Administration of Allosteric and Orthosteric Drugs to Overcome Drug-Resistance in T315I BCR-ABL1. Front. Pharmacol. 2022, 13, 862504. [Google Scholar] [CrossRef]
- Qiu, Y.; Yin, X.; Li, X.; Wang, Y.; Fu, Q.; Huang, R.; Lu, S. Untangling Dual-Targeting Therapeutic Mechanism of Epidermal Growth Factor Receptor (EGFR) Based on Reversed Allosteric Communication. Pharmaceutics 2021, 13, 747. [Google Scholar] [CrossRef]
- Ni, D.; Li, Y.; Qiu, Y.; Pu, J.; Lu, S.; Zhang, J. Combining Allosteric and Orthosteric Drugs to Overcome Drug Resistance. Trends Pharmacol. Sci. 2020, 41, 336–348. [Google Scholar] [CrossRef]
- Lu, S.; Qiu, Y.; Ni, D.; He, X.; Pu, J.; Zhang, J. Emergence of allosteric drug- resistance mutations: New challenges for allosteric drug discovery. Drug Discov. Today 2020, 25, 177–184. [Google Scholar] [CrossRef]
- Feng, L.; Lu, S.; Zheng, Z.; Chen, Y.; Zhao, Y.; Song, K.; Xue, H.; Jin, L.; Li, Y.; Huang, C.; et al. Identification of an allosteric hotspot for additive activation of PPARγ in antidiabetic effects. Sci. Bull. 2021, 66, 1559–1570. [Google Scholar] [CrossRef]
- Khan, A.; Ahsan, O.; Wei, D.-Q.; Ansari, J.K.; Najmi, M.H.; Muhammad, K.; Waheed, Y. Computational Evaluation of Abrogation of HBx-Bcl-xL Complex with High-Affinity Carbon Nanotubes (Fullerene) to Halt the Hepatitis B Virus Replication. Molecules 2021, 26, 6433. [Google Scholar] [CrossRef]
- Ramesh, P.; Shin, W.H.; Veerappapillai, S. Discovery of a potent candidate for ret-specific non-small-cell lung cancer—a combined in silico and in vitro strategy. Pharmaceutics 2021, 13, 1775. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- He, X.; Huang, N.; Qiu, Y.; Zhang, J.; Liu, Y.; Yin, X.-L.; Lu, S. Conformational Selection Mechanism Provides Structural Insights into the Optimization of APC-Asef Inhibitors. Molecules 2021, 26, 962. [Google Scholar] [CrossRef]
- Li, X.; Qi, Z.; Ni, D.; Lu, S.; Chen, L.; Chen, X. Markov State Models and Molecular Dynamics Simulations Provide Understanding of the Nucleotide-Dependent Dimerization-Based Activation of LRRK2 ROC Domain. Molecules 2021, 26, 5647. [Google Scholar] [CrossRef]
- Liang, S.; Wang, Q.; Qi, X.; Liu, Y.; Li, G.; Lu, S.; Mou, L.; Chen, X. Deciphering the Mechanism of Gilteritinib Overcoming Lorlatinib Resistance to the Double Mutant I1171N/F1174I in Anaplastic Lymphoma Kinase. Front. Cell Dev. Biol. 2021, 9, 808864. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N.long(N)method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Ye, M.; Wang, Y.; Qiu, M.; Fu, T.; Zhang, J.; Zhou, B.; Lu, S. How Parkinson’s disease-related mutations disrupt the dimerization of WD40 domain in LRRK2: A comparative molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2020, 22, 20421–20433. [Google Scholar] [CrossRef]
- Hyeon, C.; Jennings, P.A.; Adams, J.A.; Onuchic, J.N. Ligand-induced global transitions in the catalytic domain of protein kinase A. Proc. Natl. Acad. Sci. USA 2009, 106, 3023–3028. [Google Scholar] [CrossRef] [Green Version]
- Sethi, A.; Eargle, J.; Black, A.A.; Luthey-Schulten, Z. Dynamical networks in tRNA: Protein complexes. Proc. Natl. Acad. Sci. USA 2009, 106, 6620–6625. [Google Scholar] [CrossRef] [Green Version]
- Xie, T.; Yu, J.; Fu, W.; Wang, Z.; Xu, L.; Chang, S.; Wang, E.; Zhu, F.; Zeng, S.; Kang, Y.; et al. Insight into the selective binding mechanism of DNMT1 and DNMT3A inhibitors: A molecular simulation study. Phys. Chem. Chem. Phys. 2019, 21, 12931–12947. [Google Scholar] [CrossRef]
- Liu, N.; Zhou, W.; Guo, Y.; Wang, J.; Fu, W.; Sun, H.; Li, D.; Duan, M.; Hou, T. Molecular Dynamics Simulations Revealed the Regulation of Ligands to the Interactions between Androgen Receptor and Its Coactivator. J. Chem. Inf. Model. 2018, 58, 1652–1661. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Spratt, A.N.; Kannan, S.R.; Woods, L.T.; Weisman, G.A.; Quinn, T.P.; Lorson, C.L.; Sönnerborg, A.; Byrareddy, S.N.; Singh, K. Evolution, correlation, structural impact and dynamics of emerging SARS-CoV-2 variants. Comput. Struct. Biotechnol. J. 2021, 19, 3799–3809. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT | Q597A | |
|---|---|---|
| ΔGgas | −331.65 ± 15.88 | −324.55 ± 16.38 |
| ΔGsolvation | 224.89 ± 12.72 | 220.79 ± 12.82 |
| ΔGbinding | −106.76 ± 6.46 | −103.77 ± 7.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, M.; Chai, Z.; Chen, J.; Li, G.; Li, Q.; Li, M.; Ding, Y.; Lu, S.; Ju, G.; Hou, J. Insights into the Allosteric Effect of SENP1 Q597A Mutation on the Hydrolytic Reaction of SUMO1 via an Integrated Computational Study. Molecules 2022, 27, 4149. https://doi.org/10.3390/molecules27134149
Ji M, Chai Z, Chen J, Li G, Li Q, Li M, Ding Y, Lu S, Ju G, Hou J. Insights into the Allosteric Effect of SENP1 Q597A Mutation on the Hydrolytic Reaction of SUMO1 via an Integrated Computational Study. Molecules. 2022; 27(13):4149. https://doi.org/10.3390/molecules27134149
Chicago/Turabian StyleJi, Mingfei, Zongtao Chai, Jie Chen, Gang Li, Qiang Li, Miao Li, Yelei Ding, Shaoyong Lu, Guanqun Ju, and Jianquan Hou. 2022. "Insights into the Allosteric Effect of SENP1 Q597A Mutation on the Hydrolytic Reaction of SUMO1 via an Integrated Computational Study" Molecules 27, no. 13: 4149. https://doi.org/10.3390/molecules27134149
APA StyleJi, M., Chai, Z., Chen, J., Li, G., Li, Q., Li, M., Ding, Y., Lu, S., Ju, G., & Hou, J. (2022). Insights into the Allosteric Effect of SENP1 Q597A Mutation on the Hydrolytic Reaction of SUMO1 via an Integrated Computational Study. Molecules, 27(13), 4149. https://doi.org/10.3390/molecules27134149