
Potential Stereoselective Binding of Trans-(±)-Kusunokinin and Cis-(±)-Kusunokinin Isomers to CSF1R


Abstract
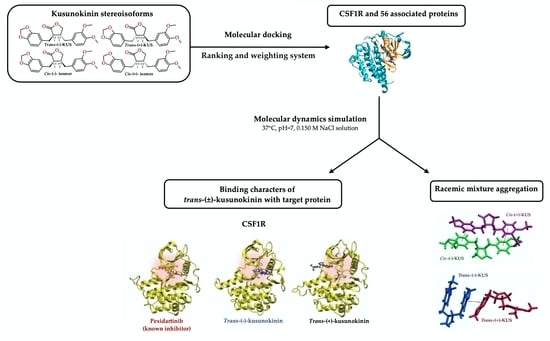
:
1. Introduction
2. Results
2.1. Target Protein(s) Screening of Four Kusunokinin Isomers among CSF1R-Related Breast Cancer Progression Proteins
2.2. Docking Score Interpretation of the Kusunokinin Isomers
2.3. Target Selection Based on Ligand-Protein Interaction Analysis
2.4. Molecular Dynamic Simulations and Relative Binding Affinities of Trans-(±)-Kusunokinin and Pexidartinib to CSF1R
2.5. Conformational Effects of Trans-(±)-Kusunokinin and Pexidartinib on CSF1R
- The ∆d value > 0 implied the residue moved further away from the protein center.
- The ∆d value < 0 implied the residue moved closer to the protein center.
- The ∆d value = 0 implied the residue stayed in the same position.
3. Discussion
4. Materials and Methods
4.1. Molecular Docking
4.1.1. Target Protein Selection and Preparation
4.1.2. Ligand Preparation
4.1.3. Docking Procedure and Binding Energy Calculation
4.1.4. Target Protein Selection for Ligand-Protein Interaction Analysis
4.1.5. Statistical Analysis
4.2. Scoring Procedure
4.3. Molecular Dynamics (MD) Simulation
4.3.1. System Preparation of the Compound-Protein Complex
4.3.2. Simulation Protocol of the Compound-Protein Complex
4.3.3. Relative Binding Free Energy Evaluation of the Compound-Protein Complex
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [CrossRef]
- Dhankhar, R.; Vyas, S.P.; Jain, A.K.; Arora, S.; Rath, G.; Goyal, A.K. Advances in novel drug delivery strategies for breast cancer therapy. Artif. Cell Blood Sub. 2010, 38, 230–249. [Google Scholar] [CrossRef] [PubMed]
- Lima, S.M.; Kehm, R.D.; Terry, M.B. Global breast cancer incidence and mortality trends by region, age-groups and fertility patterns. EClinicalMedicine 2021, 38, 100985. [Google Scholar] [CrossRef] [PubMed]
- Luque-Bolivar, A.; Pérez-Mora, E.; Villegas, V.E.; Rondón-Lagos, M. Resistance and overcoming resistance in breast cancer. Breast Cancer Targets Ther. 2020, 12, 211–229. [Google Scholar] [CrossRef]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [Green Version]
- Lau, K.; Tan, A.; Shi, Y. New and emerging targeted therapies for advanced breast cancer. Int. J. Mol. Sci. 2022, 23, 2288. [Google Scholar] [CrossRef] [PubMed]
- Masoud, V.; Pagès, G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017, 8, 120–134. [Google Scholar] [CrossRef] [Green Version]
- Lee, A. Tucatinib: First approval. Drugs 2020, 80, 1033–1038. [Google Scholar] [CrossRef]
- Moulder, S.; Borges, V.; Baetz, T.; Mcspadden, T.; Fernetich, G.; Murthy, R.; Chavira, R.; Guthrie, K.; Barrett, E.; Chia, S. Phase I study of ONT-380, a HER2 inhibitor, in patients with HER2+-advanced solid tumors, with an expansion cohort in HER2+ Metastatic Breast Cancer (MBC). Clin. Cancer Res. 2017, 23, 3529–3536. [Google Scholar] [CrossRef] [Green Version]
- Murthy, R.; Loi, S.; Okines, A.; Paplomata, E.; Hamilton, E.; Hurvitz, S.; Lin, N.; Borges, V.; Abramson, V.; Anders, C.; et al. Tucatinib, trastuzumab and capecitabine for HER2-positive metastatic breast cancer. N. Engl. J. Med. 2020, 382, 597–609. [Google Scholar] [CrossRef]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Mol. Cancer Ther. 2020, 19, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strachan, D.C.; Ruffell, B.; Oei, Y.; Bissell, M.J.; Coussens, L.M.; Pryer, N.; Daniel, D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8+ T cells. Oncoimmunology 2013, 2, e26968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, A.; Silakari, O.; Singh, R. Recent advances in colony stimulating factor-1 receptor/c-FMS as an emerging target for various therapeutic implications. Pharmacotherapy 2018, 103, 662–679. [Google Scholar] [CrossRef] [PubMed]
- Lin, C. Clinical development of colony-stimulating factor 1 receptor (CSF1R) inhibitors. J. Immunother. Precis. Oncol. 2021, 4, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Kacinski, B. CSF-1 and its receptor in breast carcinomas and neoplasms of the female reproductive tract. Mol. Reprod. Dev. 1997, 46, 71–74. [Google Scholar] [CrossRef]
- Hamilton, J. CSF-1 signal transduction. J. Leukoc. Biol. 1997, 62, 145–155. [Google Scholar] [CrossRef] [Green Version]
- Sapi, E.; Kacinski, B. The role of CSF-1 in normal and neoplastic breast physiology. Exp. Biol. Med. 1999, 220, 1–8. [Google Scholar] [CrossRef]
- Sapi, E. The role of CSF-1 in normal physiology of mammary gland and breast cancer: An update. Exp. Biol. Med. 2004, 229, 1–11. [Google Scholar] [CrossRef]
- Morandi, A.; Barbetti, V.; Riverso, M.; Dello Sbarba, P.; Rovida, E. The colony-stimulating factor-1 (CSF-1) receptor sustains ERK1/2 activation and proliferation in breast cancer cell lines. PLoS ONE 2011, 6, e27450. [Google Scholar] [CrossRef] [Green Version]
- Barbetti, V.; Morandi, A.; Tusa, I.; Digiacomo, G.; Riverso, M.; Marzi, I.; Cipolleschi, M.; Bessi, S.; Giannini, A.; Di Leo, A.; et al. Chromatin-associated CSF-1R binds to the promoter of proliferation-related genes in breast cancer cells. Oncogene 2013, 33, 4359–4364. [Google Scholar] [CrossRef] [Green Version]
- Mun, S.H.; Park, P.S.U.; Park-Min, K.H. The M-CSF receptor in osteoclasts and beyond. Exp. Mol. Med. 2020, 52, 1239–1254. [Google Scholar] [CrossRef] [PubMed]
- Kluger, H.; Dolled-Filhart, M.; Rodov, S.; Kacinski, B.; Camp, R.; Rimm, D. Macrophage colony-stimulating factor-1 receptor expression is associated with poor outcome in breast cancer by large cohort tissue microarray analysis. Clin. Cancer. Res. 2004, 10, 173–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aharinejad, S.; Salama, M.; Paulus, P.; Zins, K.; Berger, A.; Singer, C.F. Elevated CSF1 serum concentration predicts poor overall survival in women with early breast cancer. Endocr. Relat. Cancer 2013, 20, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardsen, E.; Uglehus, R.D.; Johnsen, S.H.; Busund, L. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. 2015, 35, 865–874. [Google Scholar] [PubMed]
- Mo, H.; Hao, Y.; Lv, Y.; Chen, Z.; Shen, J.; Zhou, S.; Yin, M. Overexpression of macrophage-colony stimulating factor-1 receptor as a prognostic factor for survival in cancer. Medicine 2021, 100, e25218. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Burugu, S.; Cheng, A.S.; Leung, S.C.Y.; Gao, D.; Nielsen, T.O. Prognostic significance of CSF-1R expression in early invasive breast cancer. Cancers 2021, 13, 5769. [Google Scholar] [CrossRef]
- Tap, W.; Gelderblom, H.; Palmerini, E.; Desai, J.; Bauer, S.; Blay, J.; Alcindor, T.; Ganjoo, K.; Martín-Broto, J.; Ryan, C.; et al. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): A randomised phase 3 trial. Lancet 2019, 394, 478–487. [Google Scholar] [CrossRef]
- Benner, B.; Good, L.; Quiroga, D.; Schultz, T.E.; Kassem, M.; Carson, W.E. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: A systematic review of pre-clinical and clinical development. Drug Des. Devel. Ther. 2020, 14, 1693–1704. [Google Scholar] [CrossRef]
- Cannarile, M.; Weisser, M.; Jacob, W.; Jegg, A.; Ries, C.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- El-Gamal, M.; Oh, C. Pyrrolo [3,2-c]pyridine derivatives with potential inhibitory effect against FMS kinase: In vitro biological studies. J. Enzyme Inhib. Med. Chem. 2018, 33, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, M.; Daniel, D.; Pryer, N.; Sutton, J.; Sung, V.; Wang, T.; Jeffry, U.; Oei, Y.; Kaufman, S.; Lenahan, W.; et al. Abstract 3629: BLZ945, a selective c-fms (CSF-1R) kinase inhibitor for the suppression of tumor-induced osteolytic lesions in bone. Cancer Res. 2010, 70, 3629. [Google Scholar] [CrossRef]
- Bendell, J.C.; Tolcher, A.W.; Jones, S.F.; Beeram, M.; Infante, J.R.; Larsen, P.; Rasor, K.; Garrus, J.E.; Li, J.F.; Cable, P.L.; et al. A phase 1 study of ARRY-382, an oral inhibitor of colony-stimulating factor-1 receptor (CSF1R), in patients with advanced or metastatic cancers. Mol. Cancer Ther. 2013, 12, A252. [Google Scholar] [CrossRef]
- Von Tresckow, B.; Morschhauser, F.; Ribrag, V.; Topp, M.S.; Chien, C.; Seetharam, S.; Aquino, R.; Kotoulek, S.; de Boer, C.J.; Engert, A. An open-label, multicenter, phase I/II study of JNJ-40346527, a CSF-1R inhibitor, in patients with relapsed or refractory hodgkin lymphoma. Clin. Cancer Res. 2015, 21, 1843–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genovese, M.; Hsia, E.; Belkowski, S.; Chien, C.; Masterson, T.; Thurmond, R.; Manthey, C.; Yan, X.; Ge, T.; Franks, C.; et al. Results from a phase IIA parallel group study of JNJ-40346527, an oral CSF-1R inhibitor, in patients with active rheumatoid arthritis despite disease-modifying antirheumatic drug therapy. J. Rheumatol. 2015, 42, 1752–1760. [Google Scholar] [CrossRef]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzon- Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Yıldırım, M.; Goh, K.; Cusick, M.; Barabási, A.; Vidal, M. Drug-target network. Nat. Biotechnol. 2007, 25, 1119–1126. [Google Scholar] [CrossRef]
- Dattachoudhury, S.; Sharma, R.; Kumar, A.; Jaganathan, B. Sorafenib inhibits proliferation, migration and invasion of breast cancer cells. Oncology 2020, 98, 478–486. [Google Scholar] [CrossRef]
- Sartorelli, P.; Carvalho, C.S.; Reimão, J.Q.; Lorenzi, H.; Tempone, A. Antitrypanosomal activity of a diterpene and lignans isolated from Aristolochia cymbifera. Planta Med. 2010, 76, 1454–1456. [Google Scholar] [CrossRef] [Green Version]
- Sriwiriyajan, S.; Sukpondma, Y.; Srisawat, T.; Madla, S.; Graidistm, P. (-)-Kusunokinin and piperloguminine from Piper nigrum: An alternative option to treat breast cancer. Biomed. Pharmacother. 2017, 92, 732–743. [Google Scholar] [CrossRef]
- Tedasen, A.; Dokduang, S.; Sukpondma, Y.; Lailerd, N.; Madla, S.; Sriwiriyajan, S.; Rattanaburee, T.; Tipmanee, V.; Graidist, P. (-)-Kusunokinin inhibits breast cancer in N-nitrosomethylurea-induced mammary tumor rats. Eur. J. Pharmacol. 2020, 882, 173311. [Google Scholar] [CrossRef]
- Rattanaburee, T.; Thongpanchang, T.; Wongma, K.; Tedasen, A.; Sukpondma, Y.; Graidist, P. Anticancer activity of synthetic (±)-kusunokinin and its derivative (±)-bursehernin on human cancer cell lines. Biomed. Pharmacother. 2019, 117, 109115. [Google Scholar] [CrossRef] [PubMed]
- Rattanaburee, T.; Tipmanee, V.; Tedasen, A.; Thongpanchang, T.; Graidist, P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed. Pharmacother. 2020, 129, 110361. [Google Scholar] [CrossRef] [PubMed]
- Rattanaburee, T.; Tanawattanasuntorn, T.; Thongpanchang, T.; Tipmanee, V.; Graidist, P. Trans-(−)-Kusunokinin: A potential anticancer lignan compound against HER2 in breast cancer cell lines? Molecules 2021, 26, 4537. [Google Scholar] [CrossRef] [PubMed]
- Tanawattanasuntorn, T.; Thongpanchang, T.; Rungrotmongkol, T.; Hanpaibool, C.; Graidist, P.; Tipmanee, V. (-)-Kusunokinin as a potential aldose reductase inhibitor: Equivalency observed via AKR1B1 dynamics simulation. ACS Omega 2020, 6, 606–614. [Google Scholar] [CrossRef]
- Lopes, L.M.X.; Yoshida, M.; Gottlieb, O.R. Dibenzylbutyrolactone lignans from Virola Sebifera. Phytochemistry 1983, 22, 1516–1518. [Google Scholar] [CrossRef]
- Lee, S.; Yoo, H.H.; Piao, X.L.; Kim, J.S.; Kang, S.S.; Shin, K.H. Anti-estrogenic activity of lignans from Acanthopanax chiisanensis root. Arch. Pharm. Res. 2005, 28, 186–189. [Google Scholar] [CrossRef]
- Moritani, Y.; Fukushima, C.; Ukita, T.; Miyagishima, T.; Ohmizu, H.; Iwasaki, T. Stereoselective syntheses of cis- and trans-isomers of α-Hydroxy-α,β-dibenzyl-γ-butyrolactone lignans: New syntheses of (±)-trachelogenin and (±)-guayadequiol. J. Org. Chem. 1996, 61, 6922–6930. [Google Scholar] [CrossRef]
- Veber, D.; Johnson, S.; Cheng, H.; Smith, B.; Ward, K.; Kopple, K. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Mateus, A.; Gordon, L.; Wayne, G.; Almqvist, H.; Axelsson, H.; Seashore-Ludlow, B.; Treyer, A.; Matsson, P.; Lundbäck, T.; West, A.; et al. Prediction of intracellular exposure bridges the gap between target- and cell-based drug discovery. Proc. Natl. Acad. Sci. USA 2017, 114, E6231–E6239. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Ahmad, A.; Vyawahare, A.; Khan, R. Membrane trafficking and subcellular drug targeting pathways. Front. Pharmacol. 2020, 11, 629. [Google Scholar] [CrossRef]
- Rosenkranz, A.A.; Slastnikova, T.A.; Georgiev, G.P.; Zalutsky, M.R.; Soboley, A.S. Delivery systems exploiting natural cell transport processes of macromolecules for intracellular targeting of Auger electron emitters. Nucl. Med. Biol. 2020, 80, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.; Ueno, N. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baselga, J. The EGF receptor family as targets for breast cancer therapy. Breast Cancer Res. 2000, 2, S.13. [Google Scholar] [CrossRef]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Islam, M.; Mahdi, J.; Bowen, I. Pharmacological importance of stereochemical resolution of enantiomeric drugs. Drug Saf. 1997, 17, 149–165. [Google Scholar] [CrossRef]
- Lees, P.; Hunter, R.; Reeves, P.; Toutain, P. Pharmacokinetics and pharmacodynamics of stereoisomeric drugs with particular reference to bioequivalence determination. J. Vet. Pharmacol. Ther. 2012, 35, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D. Drug disposition in three dimensions: An update on stereoselectivity in pharmacokinetics. Biopharm. Drug Dispos. 2006, 27, 387–406. [Google Scholar] [CrossRef]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Boulakirba, S.; Pfeifer, A.; Mhaidly, R.; Obba, S.; Goulard, M.; Schmitt, T.; Chaintreuil, P.; Calleja, A.; Furstoss, N.; Orange, F.; et al. IL-34 and CSF-1 display an equivalent macrophage differentiation ability but a different polarization potential. Sci. Rep. 2018, 8, 256. [Google Scholar] [CrossRef]
- Giricz, O.; Mo, Y.; Dahlman, K.B.; Cotto-Rios, X.M.; Vardabasso, C.; Nguyen, H.; Matusow, B.; Bartenstein, M.; Polishchuk, V.; Johnson, D.B.; et al. The RUNX1/IL-34/CSF-1R axis is an autocrinally regulated modulator of resistance to BRAF-V600E inhibition in melanoma. JCI Insight 2018, 3, 120422. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.W.; States, D.J. Both src-dependent and -independent mechanisms mediate phosphatidylinositol 3-kinase regulation of colony-stimulating factor 1-activated mitogen-activated protein kinases in myeloid progenitors. Mol. Cell Biol. 2000, 20, 6779–6798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, C.; Chiarini, F.; Paganelli, F.; Marmiroli, S.; Martelli, A. Crosstalks of GSK3 signaling with the mTOR network and effects on targeted therapy of cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colomiere, M.; Ward, A.; Riley, C.; Trenerry, M.; Cameron-Smith, D.; Findlay, J.; Ackland, L.; Ahmed, N. Cross talk of signals between EGFR and IL-6R through JAK2/STAT3 mediate epithelial–mesenchymal transition in ovarian carcinomas. Br. J. Cancer 2008, 100, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Khayami, R.; Hashemi, S.; Kerachian, M. Role of aldo-keto reductase family 1 member B1 (AKR1B1) in the cancer process and its therapeutic potential. J. Cell. Mol. Med. 2020, 24, 8890–8902. [Google Scholar] [CrossRef]
- Papavassiliou, K.; Papavassiliou, A. Transcription factor drug targets. J. Cell Biochem. 2016, 117, 2693–2696. [Google Scholar] [CrossRef]
- Bakheet, T.; Doig, A. Properties and identification of human protein drug targets. Bioinformatics 2009, 25, 451–457. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Han, L.; Yap, C.; Ji, Z.; Cao, Z.; Chen, Y. Therapeutic targets: Progress of their exploration and investigation of their characteristics. Pharmacol. Rev. 2006, 58, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Henley, M. Drugging fuzzy complexes in transcription. Front. Mol. Biosci. 2021, 8, 795743. [Google Scholar] [CrossRef]
- Xie, Z.; Wu, B.; Liu, Y.; Ren, W.; Tong, L.; Xiang, C.; Wei, A.; Gao, Y.; Zeng, L.; Xie, H.; et al. Novel class of colony-stimulating factor 1 receptor kinase inhibitors based on an o-aminopyridyl alkynyl scaffold as potential treatment for inflammatory disorders. J. Med. Chem. 2020, 63, 1397–1414. [Google Scholar] [CrossRef]
- Schubert, C.; Schalk-Hihi, C.; Struble, G.; Ma, H.; Petrounia, I.; Brandt, B.; Deckman, I.; Patch, R.; Player, M.; Spurlino, J.; et al. Crystal structure of the tyrosine kinase domain of colony-stimulating factor-1 receptor (cFMS) in complex with two inhibitors. J. Biol. Chem. 2007, 282, 4094–4101. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [PubMed]
- Nerurkar, S.; Dighe, S.; Williams, R. Bioequivalence of racemic drugs. J. Clin. Pharm. 1992, 32, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, G.; Perucca, E. The cost benefit ratio of enantiomeric drugs. Eur. J. Drug Metab. Pharmacokinet. 1995, 20, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Rogers, J.; Demetriades, J.; Holland, S.; Seibold, J.; Depuy, E. Pharmacokinetics and bioinversion of ibuprofen enantiomers in humans. Pharm. Res. 1994, 11, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cisneros, G.A.; Cruzeiro, V.W.D.; Duke, R.; et al. AMBER 2020; University of California: San Francisco, CA, USA, 2020. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Jewboonchu, J.; Saetang, J.; Saeloh, D.; Siriyong, T.; Rungrotmongkol, T.; Voravuthikunchai, S.P.; Tipmanee, V. Atomistic insight and modeled elucidation of conessine towards Pseudomonas aeruginosa efflux pump. J. Biomol. Struct. Dyn. 2022, 40, 1480–1489. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Name | PDB Code | Kusunokinin Isomer Docking Score 1 | Known Inhibitor | ||||
|---|---|---|---|---|---|---|---|
| Trans-(−) | Trans-(+) | Cis-(−) | Cis-(+) | Name | Docking Score | ||
| Anti-apoptosis and Survival | |||||||
| COX2 | 5IKR | −10.60 | −9.68 | −9.62 | −10.57 | Sorafenib | −9.06 |
| Hsp70 | 3ATV | −8.93 | −7.84 | −8.43 | −9.03 | Apoptozole | −8.24 |
| Hsp90a | 3O0I | −10.60 | −10.09 | −10.43 | −10.77 | Pu-H54 | −9.43 |
| Hsp90b | 3NMQ | −10.80 | −9.28 | −10.63 | −9.74 | EC44 | −10.20 |
| PAK6 | 4KS8 | −8.55 | −7.56 | −8.09 | −8.39 | Sunitinib | −7.50 |
| Survivin | 1F3H | −8.78 | −8.63 | −8.76 | −8.13 | YM155 | −7.25 |
| XIAP | 5OQW | −7.67 | −6.74 | −7.70 | −7.49 | A4E | −10.66 |
| Cell Growth and Proliferation | |||||||
| cMyc | 6G6J | −7.30 | −6.32 | −7.22 | −6.55 | MYCi975 | −6.19 |
| CDK2 | 4J52 | −9.74 | −9.14 | −9.90 | −9.90 | Dinaciclib | −8.93 |
| PLK1 | 4OYA | −8.87 | −8.87 | −8.92 | −8.84 | Pyrimidodiazepinone | −5.92 |
| ADC10 | 1C25 | −12.59 | −11.32 | −12.21 | −12.12 | SQ-22536 | −8.23 |
| AR | 1CWT | −8.50 | −8.35 | −9.36 | −9.03 | (R)-Bicalutamide | −8.55 |
| cdc25A | 4Y72 | −9.19 | −7.14 | −7.98 | −8.97 | Quinonoid | −6.90 |
| cdc25B | 1FVV | −7.62 | −7.46 | −7.63 | −7.78 | Quinonoid | −6.91 |
| CDK1 | 6P8E | −10.34 | −9.27 | −9.79 | −9.69 | CGP74514A | −10.96 |
| CDK4 | 1FVV | −8.16 | −7.22 | −7.32 | −7.55 | Palbociclib | −9.03 |
| cyclinA | 4Y72 | −8.89 | −9.56 | −8.92 | −8.69 | ligand 107 | −9.76 |
| cyclinB1 | 6P8E | −10.45 | −10.08 | −10.24 | −10.84 | Q27097368 | −9.40 |
| cyclinD1 | 1L09 | −8.18 | −7.04 | −8.20 | −8.42 | Fascaplysin | −7.57 |
| ER | 1SJ0 | −8.96 | −8.43 | −9.02 | −8.87 | E4D | −11.74 |
| GSK3b | 4JSV | −10.64 | −10.01 | −9.91 | −10.05 | Tideglusib | −8.59 |
| mTOR | 4UJA | −9.12 | −8.46 | −9.09 | −8.95 | AZD8055 | −10.21 |
| PKA | 3IW4 | −9.48 | −8.46 | −9.58 | −9.61 | AT13148 | −12.83 |
| PKC | 3LLU | −9.83 | −8.99 | −9.87 | −9.79 | Enzastaurin | −12.69 |
| RagC | 4R7H | −9.21 | −8.73 | −8.95 | −8.82 | Palomid-529 | −8.85 |
| CSF1R | 4HJO | −11.84 | −9.29 | −10.53 | −10.40 | Pexidartinib | −11.59 |
| EGFR | 5 × 02 | −9.71 | −6.45 | −9.69 | −7.14 | Erlotinib | −8.82 |
| FLT3 | 1N26 | −9.24 | −8.76 | −9.11 | −9.13 | Gilteritinib | −9.64 |
| IL6R | 6G6J | −7.74 | −7.48 | −7.61 | −7.73 | Terminolic acid | −7.11 |
| Metastasis | |||||||
| cFos | 1FOS | −5.63 | −5.07 | −5.32 | −5.51 | T-5224 | −6.20 |
| cJun | 1FOS | −5.66 | −4.95 | −5.27 | −5.51 | T-5224 | −7.72 |
| mmp12 | 4XCT | −11.19 | −10.23 | −10.71 | −10.80 | BAY-7598 | −12.47 |
| mmp9 | 5LAB | −10.41 | −10.48 | −10.37 | −9.64 | ARP101 | −12.27 |
| snail | 3W5K | −7.85 | −7.53 | −7.49 | −7.47 | Chembl4517265 | −7.70 |
| AKR1B1 | 4JIR | −11.32 | −11.15 | −11.46 | −11.59 | Epalrestat | −10.14 |
| ALP | 2GLQ | −7.26 | −7.95 | −7.36 | −7.34 | Levamisole | −6.08 |
| PALP | 3MK0 | −6.92 | −6.77 | −7.69 | −7.48 | Levamisole | −5.79 |
| TGFBR1 | 1E3G | −7.08 | −6.92 | −7.06 | −7.62 | SB431542 | −7.17 |
| TGFBR2 | 2PJY | −7.96 | −7.73 | −7.88 | −8.21 | J2V | −7.10 |
| Signaling molecules | |||||||
| AKT | 1GZN | −9.64 | −8.97 | −9.12 | −9.23 | Capivasertib | −8.50 |
| CRAF | 3OMV | −8.60 | −8.11 | −9.14 | −8.82 | Sorafenib | −9.06 |
| ERK1 | 4QTB | −9.63 | −9.24 | −9.54 | −9.65 | SCH772984 | −12.96 |
| ERK2 | 5BUJ | −9.15 | −8.65 | −9.11 | −9.42 | Q27455064 | −8.39 |
| Grb2 | 1GRI | −8.03 | −7.32 | −7.78 | −7.92 | CGP-78850 | −7.88 |
| IKK | 4KIK | −7.89 | −7.70 | −7.80 | −8.18 | Dehydrocostus Lactone | −7.28 |
| JAK1 | 5HX8 | −8.64 | −8.56 | −8.60 | −8.44 | 66P | −10.52 |
| JAK2 | 3KRR | −8.20 | −7.66 | −8.40 | −8.17 | NVP-BSK805 | −11.22 |
| JAK3 | 5TTV | −8.97 | −8.62 | −9.03 | −8.81 | Inhibitor 6 | −7.30 |
| K-RAS | 4M1W | −7.55 | −7.19 | −7.85 | −7.69 | Sotorasib | −7.11 |
| MEK1 | 2P55 | −10.34 | −9.48 | −10.63 | −10.14 | Trametinib | −13.05 |
| MEK2 | 1S9I | −10.17 | −9.41 | −10.95 | −10.06 | Trametinib | −12.17 |
| MNK2 | 6JLR | −9.86 | −9.60 | −10.02 | −9.94 | BV9 | −8.64 |
| p38 | 4MYG | −8.73 | −7.89 | −8.63 | −8.61 | PD169316 | −7.94 |
| PI3K | 3CSF | −9.47 | −8.66 | −9.12 | −9.06 | Apitolisib | −9.55 |
| PIM1 | 1YXV | −9.38 | −8.03 | −9.16 | −9.30 | 6SD | −6.95 |
| RAC1 | 1HH4 | −10.12 | −9.33 | −9.36 | −10.12 | NSC-23766 | −6.94 |
| SMAD3 | 1MK2 | −8.33 | −7.78 | −8.06 | −7.91 | SIS3 | −9.05 |
| STAT1 | 1YVL | −7.41 | −7.75 | −7.49 | −7.37 | Fludarabine | −5.41 |
| STAT3 | 6TLC | −7.84 | −7.51 | −7.63 | −7.82 | STAT3-IN-3 | −9.87 |
| STAT5 | 6MBZ | −9.52 | −9.27 | −8.78 | −8.96 | IN-2 | −8.45 |
| Target Name | Weight 1 | Kusunokinin Isomer Rank Score 2 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Trans-(−) | Trans-(+) | Cis-(−) | Cis-(+) | ||||||
| Anti-apoptosis and Survival | |||||||||
| COX2 | 2 | 4 | (8) | 2 | (4) | 1 | (2) | 3 | (6) |
| Hsp70 | 2 | 3 | (6) | 1 | (2) | 2 | (4) | 4 | (8) |
| Hsp90a | 2 | 3 | (6) | 1 | (2) | 2 | (4) | 4 | (8) |
| Hsp90b | 2 | 4 | (8) | 1 | (2) | 3 | (6) | 2 | (4) |
| PAK6 | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 3 | (6) |
| Surviving | 2 | 4 | (8) | 2 | (4) | 3 | (6) | 1 | (2) |
| XIAP | 2 | 3 | (6) | 1 | (2) | 4 | (8) | 2 | (4) |
| Sum score (weighted) 3 | 21 | (54) | 8 | (19) | |||||
| Cell growth and Proliferation | |||||||||
| cMyc | 1 | 4 | (4) | 1 | (1) | 3 | (3) | 2 | (2) |
| CDK2 | 1 | 2 | (2) | 1 | (1) | 4 | (4) | 4 | (4) |
| PLK1 | 1 | 3 | (3) | 3 | (3) | 4 | (4) | 1 | (1) |
| ADC10 | 2 | 4 | (8) | 1 | (2) | 3 | (6) | 2 | (4) |
| AR | 2 | 2 | (4) | 1 | (2) | 4 | (8) | 3 | (6) |
| cdc25A | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 3 | (6) |
| cdc25B | 2 | 2 | (4) | 1 | (2) | 3 | (6) | 4 | (8) |
| CDK1 | 2 | 4 | (8) | 1 | (2) | 3 | (6) | 2 | (4) |
| CDK4 | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 3 | (6) |
| cyclinA | 2 | 2 | (4) | 4 | (8) | 3 | (6) | 1 | (2) |
| cyclinB1 | 2 | 3 | (6) | 1 | (2) | 2 | (4) | 4 | (8) |
| cyclinD1 | 2 | 2 | (4) | 1 | (2) | 3 | (6) | 4 | (8) |
| ER | 2 | 3 | (6) | 1 | (2) | 4 | (8) | 2 | (4) |
| GSK3b | 2 | 4 | (8) | 2 | (4) | 1 | (2) | 3 | (6) |
| mTOR | 2 | 4 | (8) | 1 | (2) | 3 | (6) | 2 | (4) |
| PKA | 2 | 2 | (4) | 1 | (2) | 3 | (6) | 4 | (8) |
| PKC | 2 | 3 | (6) | 1 | (2) | 4 | (8) | 2 | (4) |
| RagC | 2 | 4 | (8) | 1 | (2) | 3 | (6) | 2 | (4) |
| CSF1R | 3 | 4 | (12) | 1 | (3) | 2 | (6) | 3 | (9) |
| EGFR | 3 | 4 | (12) | 1 | (3) | 3 | (9) | 2 | (6) |
| FLT3 | 3 | 4 | (12) | 1 | (3) | 2 | (6) | 3 | (9) |
| IL6R | 3 | 4 | (12) | 1 | (3) | 2 | (6) | 3 | (9) |
| Sum score (weighted) | 68 | (151) | 25 | (55) | 48 | (120) | 62 | (122) | |
| Metastasis | |||||||||
| cFos | 1 | 4 | (4) | 1 | (1) | 2 | (2) | 3 | (3) |
| cJun | 1 | 4 | (4) | 1 | (1) | 2 | (2) | 3 | (3) |
| mmp12 | 1 | 4 | (4) | 1 | (1) | 2 | (2) | 3 | (3) |
| mmp9 | 1 | 3 | (3) | 4 | (4) | 2 | (2) | 1 | (1) |
| Snail | 1 | 4 | (4) | 3 | (3) | 2 | (2) | 1 | (1) |
| AKR1B1 | 2 | 2 | (4) | 1 | (2) | 3 | (6) | 4 | (8) |
| ALP | 2 | 1 | (2) | 4 | (8) | 3 | (6) | 2 | (4) |
| PALP | 2 | 2 | (4) | 1 | (2) | 4 | (8) | 3 | (6) |
| TGFBR1 | 3 | 3 | (9) | 1 | (3) | 2 | (6) | 4 | (12) |
| TGFBR2 | 3 | 3 | (9) | 1 | (3) | 2 | (6) | 4 | (12) |
| Sum score (weighted) | 33 | (52) | 19 | (30) | 28 | (45) | 29 | (56) | |
| Signaling molecules | |||||||||
| AKT | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 3 | (6) |
| CRAF | 2 | 2 | (4) | 1 | (2) | 4 | (8) | 3 | (6) |
| ERK1 | 2 | 3 | (6) | 1 | (2) | 2 | (4) | 4 | (8) |
| ERK2 | 2 | 3 | (6) | 1 | (2) | 2 | (4) | 4 | (8) |
| Grb2 | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 3 | (6) |
| IKK | 2 | 3 | (6) | 1 | (2) | 2 | (4) | 4 | (8) |
| JAK1 | 2 | 4 | (8) | 2 | (4) | 3 | (6) | 1 | (2) |
| JAK2 | 2 | 3 | (6) | 1 | (2) | 4 | (8) | 2 | (4) |
| JAK3 | 2 | 3 | (6) | 1 | (2) | 4 | (8) | 2 | (4) |
| K-RAS | 2 | 2 | (4) | 1 | (2) | 4 | (8) | 3 | (6) |
| MEK1 | 2 | 3 | (6) | 1 | (2) | 4 | (8) | 2 | (4) |
| MEK2 | 2 | 3 | (6) | 1 | (2) | 4 | (8) | 2 | (4) |
| MNK2 | 2 | 2 | (4) | 1 | (2) | 4 | (8) | 3 | (6) |
| p38 | 2 | 2 | (4) | 4 | (8) | 3 | (6) | 1 | (2) |
| PI3K | 2 | 4 | (8) | 1 | (2) | 3 | (6) | 2 | (4) |
| PIM1 | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 3 | (6) |
| RAC1 | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 4 | (8) |
| SMAD3 | 2 | 4 | (8) | 1 | (2) | 3 | (6) | 2 | (4) |
| STAT1 | 2 | 2 | (4) | 4 | (8) | 3 | (6) | 1 | (2) |
| STAT3 | 2 | 4 | (8) | 1 | (2) | 2 | (4) | 3 | (6) |
| STAT5 | 2 | 4 | (8) | 3 | (6) | 1 | (2) | 2 | (4) |
| Sum score (weighted) | 71 | (148) | 57 | (114) | 61 | (122) | 32 | (64) | |
| Target and Interaction | Compounds | ||||
|---|---|---|---|---|---|
| Trans-(−) | Trans-(+) | Cis-(−) | Cis-(+) | Inhibitor 1 | |
| CSF1R | |||||
| π-π stacking | Trp550 | Phe797 | Trp550 | Tyr665 | Trp550 |
| Hydrogen bond | Tyr546 Arg549 Trp550 | Thr663 Cys666 Asp796 | Arg549 Trp550 Thr663 | Thr663 Tyr665 Asp796 | Arg459 Trp550 Glu664 Cys666 |
| EGFR | |||||
| Hydrogen bond | Lys745 Asp855 Phe856 | Lys745 Asp855 Phe856 | Lys745 Phe856 | Lys745 Asp855 Phe856 | Asp855 Phe856 |
| Ligand | MM/GBSA (kcal/mol) | MM/PBSA (kcal/mol) |
|---|---|---|
| Trans-(+)-kusunokinin | −22.00 ± 0.14 | 6.92 ± 0.14 |
| Trans-(−)-kusunokinin | −54.67 ± 0.09 | 0.34 ± 0.15 |
| Pexidartinib | −61.78 ± 0.09 | −7.27 ± 0.14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chompunud Na Ayudhya, C.; Graidist, P.; Tipmanee, V. Potential Stereoselective Binding of Trans-(±)-Kusunokinin and Cis-(±)-Kusunokinin Isomers to CSF1R. Molecules 2022, 27, 4194. https://doi.org/10.3390/molecules27134194
Chompunud Na Ayudhya C, Graidist P, Tipmanee V. Potential Stereoselective Binding of Trans-(±)-Kusunokinin and Cis-(±)-Kusunokinin Isomers to CSF1R. Molecules. 2022; 27(13):4194. https://doi.org/10.3390/molecules27134194
Chicago/Turabian StyleChompunud Na Ayudhya, Chompunud, Potchanapond Graidist, and Varomyalin Tipmanee. 2022. "Potential Stereoselective Binding of Trans-(±)-Kusunokinin and Cis-(±)-Kusunokinin Isomers to CSF1R" Molecules 27, no. 13: 4194. https://doi.org/10.3390/molecules27134194
APA StyleChompunud Na Ayudhya, C., Graidist, P., & Tipmanee, V. (2022). Potential Stereoselective Binding of Trans-(±)-Kusunokinin and Cis-(±)-Kusunokinin Isomers to CSF1R. Molecules, 27(13), 4194. https://doi.org/10.3390/molecules27134194