
The Nucleoside/Nucleotide Analogs Tenofovir and Emtricitabine Are Inactive against SARS-CoV-2

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. TAF, TDF, TFV, and FTC Are Inactive against SARS-CoV-2 in Cell-Based Assays
2.2. Efficient Formation of Active Metabolites Are Observed in A549 Cells
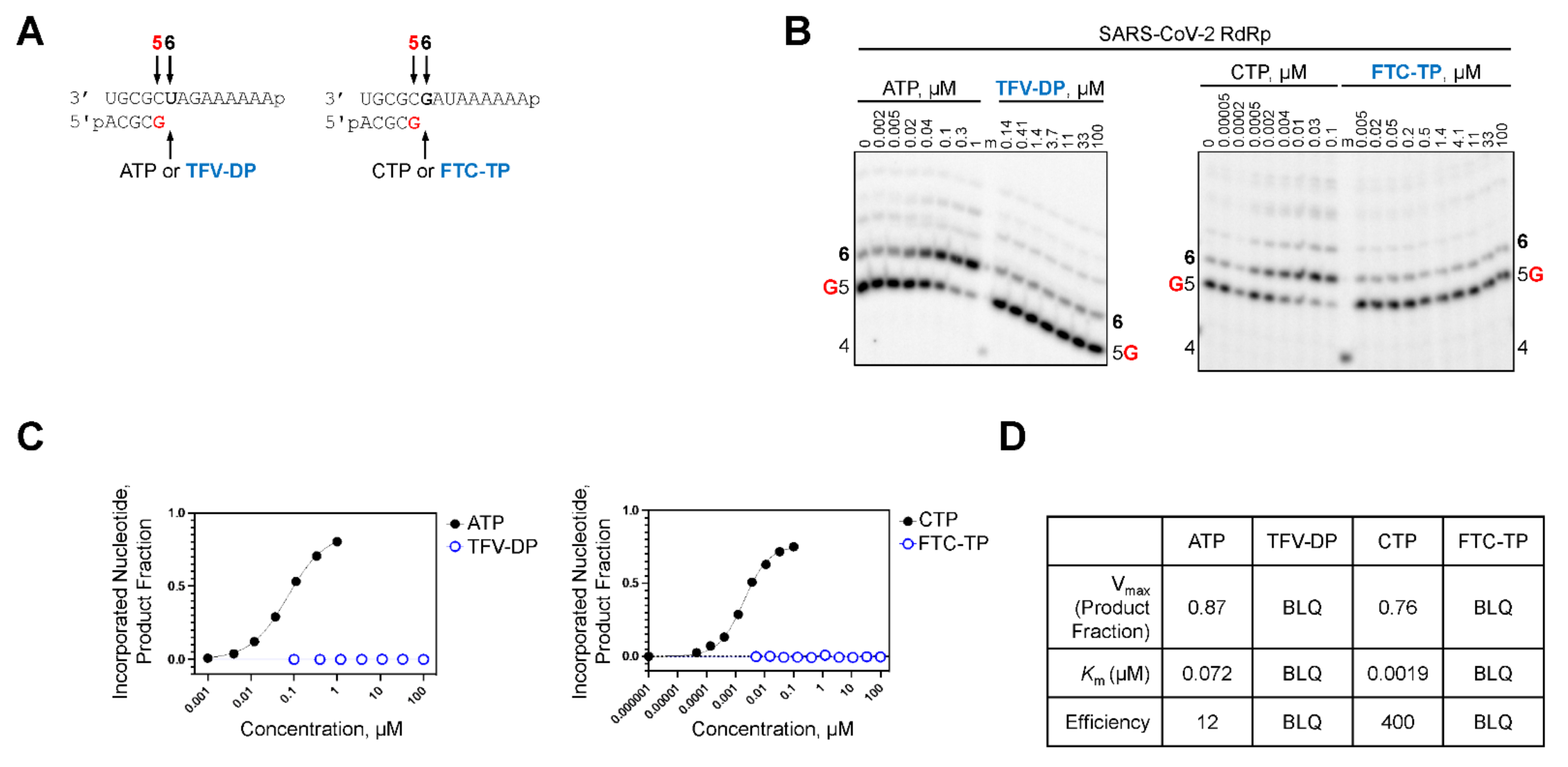
2.3. TFV-DP and FTC-TP Are Poor Substrates of SARS-CoV-2 RdRp
2.4. TFV-DP and FTC-TP Do Not Inhibit RNA Synthesis by SARS-CoV-2 RdRp
2.5. TFV-DP and FTC-TP Do Not Fit Well into the SARS-CoV-2 RdRp Active Site
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Antiviral and Cytoxicity Evaluation in A549-hACE2 Cells
4.3. Evaluation of Antiviral Assays in NHBE Cells
4.4. Measurement of Active Metabolites in Cells
4.5. Biochemical Assays
4.5.1. SARS-CoV-2 RdRp Expression and Purification
4.5.2. Selective Incorporation of Nucleotide Analogs
4.5.3. Inhibition of SARS-CoV-2 RdRp-Catalyzed RNA Synthesis by Nucleotide Analogs in a Gel-Based Assay
4.5.4. Inhibition of SARS-CoV-2 RdRp in a Filter-Binding Assay
4.6. Structural Aanalysis
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Graham, R.L.; Donaldson, E.F.; Baric, R.S. A decade after SARS: Strategies for controlling emerging coronaviruses. Nat. Rev. Microbiol. 2013, 11, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copertino, D.C., Jr.; Casado Lima, B.C.; Duarte, R.R.R.; Powell, T.R.; Ormsby, C.E.; Wilkin, T.; Gulick, R.M.; de Mulder Rougvie, M.; Nixon, D.F. Antiretroviral drug activity and potential for pre-exposure prophylaxis against COVID-19 and HIV infection. J. Biomol. Struct. Dyn. 2021, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2021, 39, 3204–3212. [Google Scholar] [CrossRef] [PubMed]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef]
- Jockusch, S.; Tao, C.; Li, X.; Anderson, T.K.; Chien, M.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Triphosphates of the Two Components in DESCOVY and TRUVADA are Inhibitors of the SARS-CoV-2 Polymerase [Pre-Print]. BioRxiv. 2020. [Google Scholar]
- Western Cape Department of Health in collaboration with the National Institute for Communicable Diseases, South Africa. Risk Factors for Coronavirus Disease 2019 (COVID-19) Death in a Population Cohort Study from the Western Cape Province, South Africa. Clin. Infect. Dis. 2021, 73, e2005–e2015. [Google Scholar] [CrossRef]
- Ssentongo, P.; Heilbrunn, E.S.; Ssentongo, A.E.; Advani, S.; Chinchilli, V.M.; Nunez, J.J.; Du, P. Epidemiology and outcomes of COVID-19 in HIV-infected individuals: A systematic review and meta-analysis. Sci. Rep. 2021, 11, 6283. [Google Scholar] [CrossRef]
- Callebaut, C.; Liu, Y.; Babusis, D.; Ray, A.; Miller, M.; Kitrinos, K. Viability of primary osteoblasts after treatment with tenofovir alafenamide: Lack of cytotoxicity at clinically relevant drug concentrations. PLoS ONE 2017, 12, e0169948. [Google Scholar] [CrossRef]
- Wang, L.H.; Begley, J.; St. Claire, R.L.; Harris, J.; Wakeford, C.; Rousseau, F.S. Pharmacokinetic and pharmacodynamic characteristics of emtricitabine support its once daily dosing for the treatment of HIV infection. AIDS Res. Hum. Retrovir. 2004, 20, 1173–1182. [Google Scholar] [CrossRef]
- Borroto-Esoda, K.; Vela, J.E.; Myrick, F.; Ray, A.S.; Miller, M.D. In vitro evaluation of the anti-HIV activity and metabolic interactions of tenofovir and emtricitabine. Antivir. Ther. 2006, 11, 377–384. [Google Scholar] [CrossRef]
- Stray, K.M.; Park, Y.; Babusis, D.; Callebaut, C.; Cihlar, T.; Ray, A.S.; Perron, M. Tenofovir alafenamide (TAF) does not deplete mitochondrial DNA in human T-cell lines at intracellular concentrations exceeding clinically relevant drug exposures. Antivir. Res. 2017, 140, 116–120. [Google Scholar] [CrossRef]
- Li, R.; Liclican, A.; Xu, Y.; Pitts, J.; Niu, C.; Zhang, J.; Kim, C.; Zhao, X.; Soohoo, D.; Babusis, D.; et al. Key Metabolic Enzymes Involved in Remdesivir Activation in Human Lung Cells. Antimicrob. Agents Chemother. 2021, 65, e0060221. [Google Scholar] [CrossRef]
- Jiang, R.D.; Shen, H.; Piao, Y.J. The morphometrical analysis on the ultrastructure of A549 cells. Rom. J. Morphol. Embryol. 2010, 51, 663–667. [Google Scholar]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Remdesivir is a direct-acting antiviral that inhibits RNA dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.J.; Lee, H.W.; Tchesnokov, E.P.; Perry, J.K.; Feng, J.Y.; Bilello, J.P.; Porter, D.P.; Gotte, M. Efficient incorporation and template-dependent polymerase inhibition are major determinants for the broad-spectrum antiviral activity of remdesivir. J. Biol. Chem. 2022, 298, 101529. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Gordon, C.J.; Woolner, E.; Kocinkova, D.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Gotte, M. Template-dependent inhibition of coronavirus RNA-dependent RNA polymerase by remdesivir reveals a second mechanism of action. J. Biol. Chem. 2020, 295, 16156–16165. [Google Scholar] [CrossRef]
- Stevens, L.J.; Pruijssers, A.J.; Lee, H.W.; Gordon, C.J.; Tchesnokov, E.P.; Gribble, J.; George, A.S.; Hughes, T.M.; Lu, X.; Li, J.; et al. Mutations in the SARS-CoV-2 RNA dependent RNA polymerase confer resistance to remdesivir by distinct mechanisms. Sci. Transl. Med. 2022, eabo0718. [Google Scholar] [CrossRef]
- Appleby, T.C.; Perry, J.K.; Murakami, E.; Barauskas, O.; Feng, J.; Cho, A.; Fox, D., 3rd; Wetmore, D.R.; McGrath, M.E.; Ray, A.S.; et al. Viral replication. Struct. Basis RNA Replication Hepat. C Virus Polym. Sci. 2015, 347, 771–775. [Google Scholar]
- Gong, P.; Peersen, O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2010, 107, 22505–22510. [Google Scholar] [CrossRef] [Green Version]
- Zamyatkin, D.F.; Parra, F.; Alonso, J.M.; Harki, D.A.; Peterson, B.R.; Grochulski, P.; Ng, K.K. Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. J. Biol. Chem. 2008, 283, 7705–7712. [Google Scholar] [CrossRef] [Green Version]
- Deval, J.; Symons, J.A.; Beigelman, L. Inhibition of viral RNA polymerases by nucleoside and nucleotide analogs: Therapeutic applications against positive-strand RNA viruses beyond hepatitis C virus. Curr. Opin. Virol. 2014, 9, 1–7. [Google Scholar] [CrossRef]
- Krausslich, H.G.; Bartenschlager, R. Antiviral Strategies; Springer: Berlin/Heidelberg, Germany, 2009; Volume 189. [Google Scholar]
- Jordan, P.C.; Stevens, S.K.; Deval, J. Nucleosides for the treatment of respiratory RNA virus infections. Antivir. Chem. Chemother. 2018, 26, 2040206618764483. [Google Scholar] [CrossRef]
- Rocha-Pereira, J.; Jochmans, D.; Debing, Y.; Verbeken, E.; Nascimento, M.S.; Neyts, J. The viral polymerase inhibitor 2’-C-methylcytidine inhibits Norwalk virus replication and protects against norovirus-induced diarrhea and mortality in a mouse model. J. Virol. 2013, 87, 11798–11805. [Google Scholar] [CrossRef] [Green Version]
- Waters, L.; Rockstroh, J.K. Antiretroviral HIV drugs in COVID-19 research: Promises and risks. An opinion piece. HIV Med. 2020. [Google Scholar] [CrossRef]
- Mossel, E.C.; Huang, C.; Narayanan, K.; Makino, S.; Tesh, R.B.; Peters, C.J. Exogenous ACE2 expression allows refractory cell lines to support severe acute respiratory syndrome coronavirus replication. J. Virol. 2005, 79, 3846–3850. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.M.; Wolf, J.D.; Lieber, C.M.; Sourimant, J.; Lin, M.J.; Babusis, D.; DuPont, V.; Chan, J.; Barrett, K.T.; Lye, D.; et al. Oral prodrug of remdesivir parent GS-441524 is efficacious against SARS-CoV-2 in ferrets. Nat. Commun. 2021, 12, 6415. [Google Scholar] [CrossRef]
- Xie, X.; Muruato, A.E.; Zhang, X.; Lokugamage, K.G.; Fontes-Garfias, C.R.; Zou, J.; Liu, J.; Ren, P.; Balakrishnan, M.; Cihlar, T.; et al. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat. Commun. 2020, 11, 5214. [Google Scholar] [CrossRef]
- Schafer, A.; Martinez, D.R.; Won, J.J.; Meganck, R.M.; Moreira, F.R.; Brown, A.J.; Gully, K.L.; Zweigart, M.R.; Conrad, W.S.; May, S.R.; et al. Therapeutic treatment with an oral prodrug of the remdesivir parental nucleoside is protective against SARS-CoV-2 pathogenesis in mice. Sci. Transl. Med. 2022, 14, eabm3410. [Google Scholar] [CrossRef]
- Berger, I.; Fitzgerald, D.J.; Richmond, T.J. Baculovirus expression system for heterologous multiprotein complexes. Nat. Biotechnol. 2004, 22, 1583–1587. [Google Scholar] [CrossRef]
- Bieniossek, C.; Richmond, T.J.; Berger, I. MultiBac: Multigene baculovirus-based eukaryotic protein complex production. Curr. Protoc. Protein Sci. 2008, 51, 5–20. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. The Antiviral Compound Remdesivir Potently Inhibits RNA-Dependent RNA Polymerase from Middle East Respiratory Syndrome Coronavirus. J. Biol. Chem. 2020, 295, 4773–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, C.J.; Tchesnokov, E.P.; Schinazi, R.F.; Gotte, M. Molnupiravir promotes SARS-CoV-2 mutagenesis via the RNA template. J. Biol. Chem. 2021, 297, 100770. [Google Scholar] [CrossRef] [PubMed]
- Tchesnokov, E.P.; Raeisimakiani, P.; Ngure, M.; Marchant, D.; Gotte, M. Recombinant RNA-Dependent RNA Polymerase Complex of Ebola Virus. Sci. Rep. 2018, 8, 3970. [Google Scholar] [CrossRef] [PubMed]
- Tchesnokov, E.P.; Feng, J.Y.; Porter, D.P.; Gotte, M. Mechanism of Inhibition of Ebola Virus RNA-Dependent RNA Polymerase by Remdesivir. Viruses 2019, 11, 326. [Google Scholar] [CrossRef] [Green Version]
- Barauskas, O.; Xing, W.; Aguayo, E.; Willkom, M.; Sapre, A.; Clarke, M.; Birkus, G.; Schultz, B.E.; Sakowicz, R.; Kwon, H.; et al. Biochemical characterization of recombinant influenza A polymerase heterotrimer complex: Polymerase activity and mechanisms of action of nucleotide analogs. PLoS ONE 2017, 12, e0185998. [Google Scholar] [CrossRef]
- Chen, J.; Malone, B.; Llewellyn, E.; Grasso, M.; Shelton, P.M.M.; Olinares, P.D.B.; Maruthi, K.; Eng, E.T.; Vatandaslar, H.; Chait, B.T.; et al. Structural Basis for Helicase-Polymerase Coupling in the SARS-CoV-2 Replication-Transcription Complex. Cell 2020, 182, 1560–1573.e13. [Google Scholar] [CrossRef]
- Tuske, S.; Sarafianos, S.G.; Clark, A.D., Jr.; Ding, J.; Naeger, L.K.; White, K.L.; Miller, M.D.; Gibbs, C.S.; Boyer, P.L.; Clark, P.; et al. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat. Struct. Mol. Biol. 2004, 11, 469–474. [Google Scholar] [CrossRef]
- Hung, M.; Tokarsky, E.J.; Lagpacan, L.; Zhang, L.; Suo, Z.; Lansdon, E.B. Elucidating molecular interactions of L-nucleotides with HIV-1 reverse transcriptase and mechanism of M184V-caused drug resistance. Commun. Biol. 2019, 2, 469. [Google Scholar] [CrossRef] [Green Version]
- Lansdon, E.B.; Samuel, D.; Lagpacan, L.; Brendza, K.M.; White, K.L.; Hung, M.; Liu, X.; Boojamra, C.G.; Mackman, R.L.; Cihlar, T.; et al. Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA complex. J. Mol. Biol. 2010, 397, 967–978. [Google Scholar] [CrossRef]
- Bertoletti, N.; Chan, A.H.; Schinazi, R.F.; Yin, Y.W.; Anderson, K.S. Structural insights into the recognition of nucleoside reverse transcriptase inhibitors by HIV-1 reverse transcriptase: First crystal structures with reverse transcriptase and the active triphosphate forms of lamivudine and emtricitabine. Protein Sci. 2019, 28, 1664–1675. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Cell-Based Antiviral Assay EC50 (μM) | Clinical Drug Exposure and Active Metabolite Levels in PBMC | ||
|---|---|---|---|---|
| Nucleoside/Tide Analogs | A549-hACE2 NanoLuc Reporter 1 | NHBE Firefly Luc Reporter 2 | Drug Exposure (μM) (Active Metabolite) 3 | Plasma Protein Binding (%) |
| RDV | 0.104 ± 0.016 | 0.037 ± 0.017 | Cmax = 7.3, 3.7 ** (10.2 μM in PBMC) | 88 |
| TAF | >50 | >50 | Cmax = 0.4 μM | 80 |
| TDF | >50 | >50 | Not detectable | - |
| TFV | >500 | Not done | NA 4 | - |
| FTC | >50 | >50 | Cmax = 7.9 μM | 4 |
| Nucleoside/Tide Analogs | Active Metabolite Formed in Cell Culture (μM) in A549 at 24-h | Extrapolated Active Metabolite under EC50 (μM) 1 in A549 at 24-h | Metabolite Level under Clinical Exposure (μM) 2 Post Multiple Doses |
|---|---|---|---|
| RDV | 21.0 ± 2.2 (with 1 μM RDV) | 1.4 | 10.2 (with RDV 200/100 mg QD) |
| TAF | 126 ± 50 (with 1 μM TAF) | >6300 | 1 (with TAF 25 mg QD) |
| TDF | 14 ± 2 (with 1 μM TDV) | >700 | 0.2 (with TDF 300 mg QD) |
| TFV | 1.7 ± 0.6 (with 10 μM TFV) | NA3 | NA 3 |
| FTC | 5.8 ± 1.8 (with 1 μM FTC) | >290 | 20 (with FTC 200 mg QD) |
| Nucleoside Analogs or Nucleotide Prodrugs | NTP or Active Metabolites | SARS-CoV-2 RNA Polymerase Assay 1 | |||
|---|---|---|---|---|---|
| Vmax (Product Fraction) 2 | Km (μM) | Incorporation Efficiency (Vmax/Km) | Efficiency Relative to the Natural NTP Substrate | ||
| Adenosine | ATP | 0.87 | 0.072 | 12 | - |
| RDV | GS-443902 | 0.74 | 0.0089 | 83 | 3.6-fold higher than ATP 3 |
| TAF, TDF, TFV | TFV-DP | BLQ 4 | BLQ 4 | BLQ 4 | Too low to be measured |
| Cytidine | CTP | 0.76 | 0.019 | 400 | - |
| FTC | FTC-TP | BLQ 4 | BLQ 4 | BLQ 4 | Too low to be measured |
| Nucleoside Analogs or Nucleotide Prodrugs | NTP or Active Metabolites | HIV-1 RT DNA Polymerase Assay 1 | |||
| Vmax | Km (μM) | Incorporation Efficiency (Vmax/Km) | Efficiency Relative to the Natural dNTP Substrate | ||
| 2′-deoxyadenosine | dATP | 0.87 | 0.0019 | 458 | - |
| TAF, TDF, TFV | TFV-DP | 0.81 | 0.0032 | 253 | 1.5-fold lower than dATP |
| 2′-deoxycytidine | dCTP | 0.90 | 0.0027 | 333 | - |
| FTC | FTC-TP | 0.84 | 0.0051 | 165 | 2-fold lower than dCTP |
| Nucleoside Analogs or Nucleotide Prodrugs | Active Metabolites | Inhibition of SARS-CoV-2 RdRp IC50 (μM) 1 |
|---|---|---|
| TAF, TDF, TFV | TFV-DP | >100 |
| FTC | FTC-TP | >100 |
| RDV (positive control) | RDV-TP | 1.0 ± 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, J.Y.; Du Pont, V.; Babusis, D.; Gordon, C.J.; Tchesnokov, E.P.; Perry, J.K.; Duong, V.; Vijjapurapu, A.; Zhao, X.; Chan, J.; et al. The Nucleoside/Nucleotide Analogs Tenofovir and Emtricitabine Are Inactive against SARS-CoV-2. Molecules 2022, 27, 4212. https://doi.org/10.3390/molecules27134212
Feng JY, Du Pont V, Babusis D, Gordon CJ, Tchesnokov EP, Perry JK, Duong V, Vijjapurapu A, Zhao X, Chan J, et al. The Nucleoside/Nucleotide Analogs Tenofovir and Emtricitabine Are Inactive against SARS-CoV-2. Molecules. 2022; 27(13):4212. https://doi.org/10.3390/molecules27134212
Chicago/Turabian StyleFeng, Joy Y., Venice Du Pont, Darius Babusis, Calvin J. Gordon, Egor P. Tchesnokov, Jason K. Perry, Vincent Duong, Arya Vijjapurapu, Xiaofeng Zhao, Julie Chan, and et al. 2022. "The Nucleoside/Nucleotide Analogs Tenofovir and Emtricitabine Are Inactive against SARS-CoV-2" Molecules 27, no. 13: 4212. https://doi.org/10.3390/molecules27134212
APA StyleFeng, J. Y., Du Pont, V., Babusis, D., Gordon, C. J., Tchesnokov, E. P., Perry, J. K., Duong, V., Vijjapurapu, A., Zhao, X., Chan, J., Cohen, C., Juneja, K., Cihlar, T., Götte, M., & Bilello, J. P. (2022). The Nucleoside/Nucleotide Analogs Tenofovir and Emtricitabine Are Inactive against SARS-CoV-2. Molecules, 27(13), 4212. https://doi.org/10.3390/molecules27134212